

The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis
- PMID: 29616220
- PMCID: PMC5869935
- DOI: 10.3389/fmed.2018.00043
The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis
Abstract
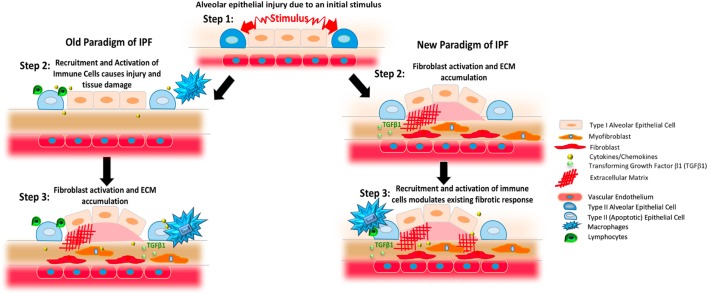
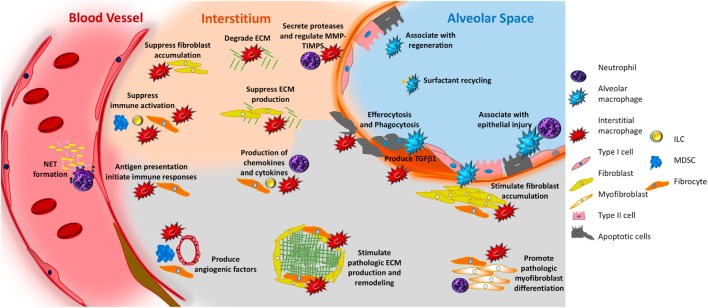
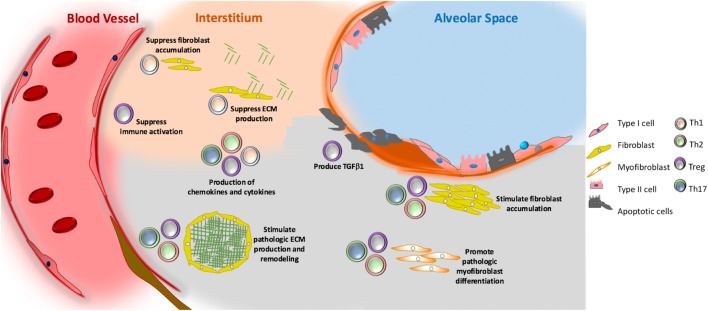
The contribution of the immune system to idiopathic pulmonary fibrosis (IPF) remains poorly understood. While most sources agree that IPF does not result from a primary immunopathogenic mechanism, evidence gleaned from animal modeling and human studies suggests that innate and adaptive immune processes can orchestrate existing fibrotic responses. This review will synthesize the available data regarding the complex role of professional immune cells in IPF. The role of innate immune populations such as monocytes, macrophages, myeloid suppressor cells, and innate lymphoid cells will be discussed, as will the activation of these cells via pathogen-associated molecular patterns derived from invading or commensural microbes, and danger-associated molecular patterns derived from injured cells and tissues. The contribution of adaptive immune responses driven by T-helper cells and B cells will be reviewed as well. Each form of immune activation will be discussed in the context of its relationship to environmental and genetic factors, disease outcomes, and potential therapies. We conclude with discussion of unanswered questions and opportunities for future study in this area.
Keywords: adaptive immunity; fibroproliferation; innate immunity; lymphocyte; macrophage.
Figures
Similar articles
-
Evolving Perspectives on Innate Immune Mechanisms of IPF.Front Mol Biosci. 2021 Aug 9;8:676569. doi: 10.3389/fmolb.2021.676569. eCollection 2021. Front Mol Biosci. 2021. PMID: 34434962 Free PMC article. Review.
-
Dendritic Cells Are the Intriguing Players in the Puzzle of Idiopathic Pulmonary Fibrosis Pathogenesis.Front Immunol. 2021 Apr 30;12:664109. doi: 10.3389/fimmu.2021.664109. eCollection 2021. Front Immunol. 2021. PMID: 33995394 Free PMC article. Review.
-
Inflammation and immunity in IPF pathogenesis and treatment.Respir Med. 2019 Feb;147:79-91. doi: 10.1016/j.rmed.2018.12.015. Epub 2019 Jan 9. Respir Med. 2019. PMID: 30704705 Review.
-
T cells in idiopathic pulmonary fibrosis: crucial but controversial.Cell Death Discov. 2023 Feb 14;9(1):62. doi: 10.1038/s41420-023-01344-x. Cell Death Discov. 2023. PMID: 36788232 Free PMC article. Review.
-
Caveolin scaffolding domain (CSD) peptide LTI-2355 modulates the phagocytic and synthetic activity of lung derived myeloid cells in Idiopathic Pulmonary Fibrosis (IPF) and Post-acute sequelae of COVID-fibrosis (PASC-F).bioRxiv [Preprint]. 2024 Jan 16:2023.12.01.569608. doi: 10.1101/2023.12.01.569608. bioRxiv. 2024. PMID: 38654821 Free PMC article. Preprint.
Cited by
-
Quantitative Proteomic Analysis in Alveolar Type II Cells Reveals the Different Capacities of RAS and TGF-β to Induce Epithelial-Mesenchymal Transition.Front Mol Biosci. 2021 Mar 19;8:595712. doi: 10.3389/fmolb.2021.595712. eCollection 2021. Front Mol Biosci. 2021. PMID: 33869273 Free PMC article.
-
Modelling bronchial epithelial-fibroblast cross-talk in idiopathic pulmonary fibrosis (IPF) using a human-derived in vitro air liquid interface (ALI) culture.Sci Rep. 2024 Jan 2;14(1):240. doi: 10.1038/s41598-023-50618-y. Sci Rep. 2024. PMID: 38168149 Free PMC article.
-
Construction of an artificial neural network diagnostic model and investigation of immune cell infiltration characteristics for idiopathic pulmonary fibrosis.BMC Pulm Med. 2024 Sep 17;24(1):458. doi: 10.1186/s12890-024-03249-6. BMC Pulm Med. 2024. PMID: 39289672 Free PMC article.
-
Classical monocyte-derived macrophages as therapeutic targets of umbilical cord mesenchymal stem cells: comparison of intratracheal and intravenous administration in a mouse model of pulmonary fibrosis.Respir Res. 2023 Mar 5;24(1):68. doi: 10.1186/s12931-023-02357-x. Respir Res. 2023. PMID: 36870972 Free PMC article.
-
Management of BMI Is a Potential New Approach for the Prevention of Idiopathic Pulmonary Fibrosis.Front Genet. 2022 Mar 11;13:821029. doi: 10.3389/fgene.2022.821029. eCollection 2022. Front Genet. 2022. PMID: 35360873 Free PMC article.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources