

Antineoplastic Drug-Induced Cardiotoxicity: A Redox Perspective
- PMID: 29563880
- PMCID: PMC5846016
- DOI: 10.3389/fphys.2018.00167
Antineoplastic Drug-Induced Cardiotoxicity: A Redox Perspective
Abstract
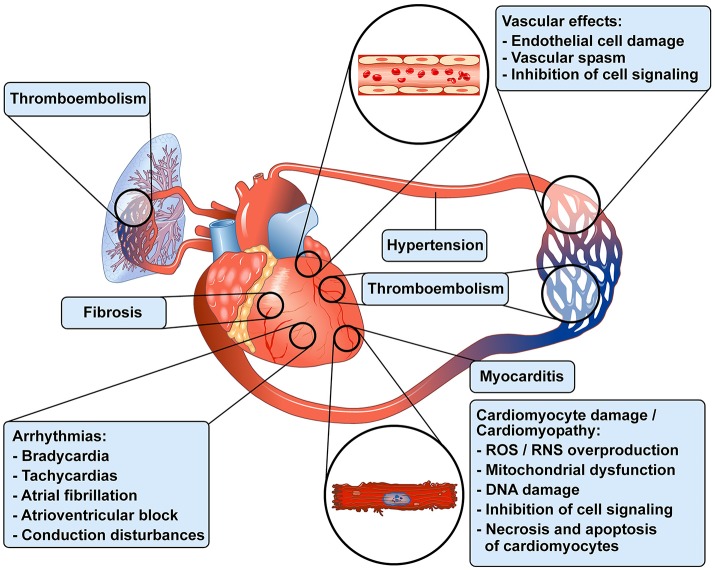
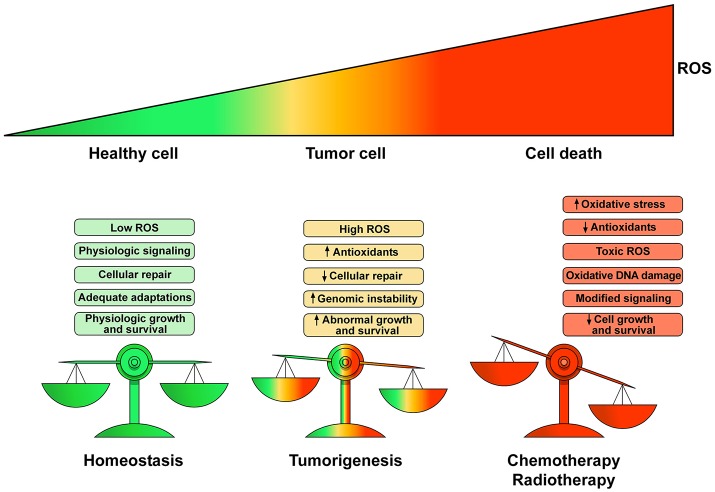
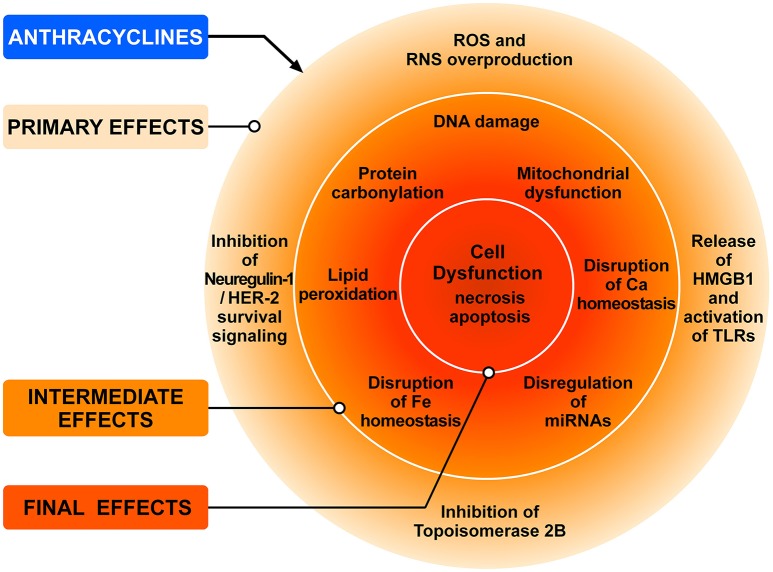
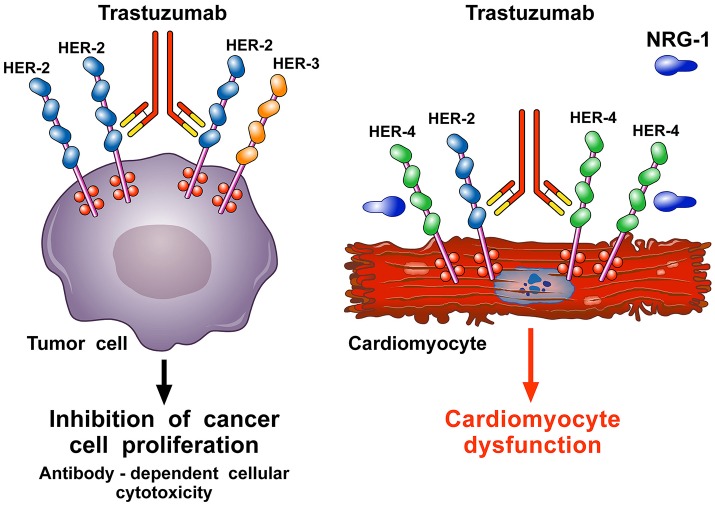
Antineoplastic drugs can be associated with several side effects, including cardiovascular toxicity (CTX). Biochemical studies have identified multiple mechanisms of CTX. Chemoterapeutic agents can alter redox homeostasis by increasing the production of reactive oxygen species (ROS) and reactive nitrogen species RNS. Cellular sources of ROS/RNS are cardiomyocytes, endothelial cells, stromal and inflammatory cells in the heart. Mitochondria, peroxisomes and other subcellular components are central hubs that control redox homeostasis. Mitochondria are central targets for antineoplastic drug-induced CTX. Understanding the mechanisms of CTX is fundamental for effective cardioprotection, without compromising the efficacy of anticancer treatments. Type 1 CTX is associated with irreversible cardiac cell injury and is typically caused by anthracyclines and conventional chemotherapeutic agents. Type 2 CTX, associated with reversible myocardial dysfunction, is generally caused by biologicals and targeted drugs. Although oxidative/nitrosative reactions play a central role in CTX caused by different antineoplastic drugs, additional mechanisms involving directly and indirectly cardiomyocytes and inflammatory cells play a role in cardiovascular toxicities. Identification of cardiologic risk factors and an integrated approach using molecular, imaging, and clinical data may allow the selection of patients at risk of developing chemotherapy-related CTX. Although the last decade has witnessed intense research related to the molecular and biochemical mechanisms of CTX of antineoplastic drugs, experimental and clinical studies are urgently needed to balance safety and efficacy of novel cancer therapies.
Keywords: HER-2 inhibitors; chemotherapy; oxidative/nitrosative stress; tyrosine kinase inhibitors; vascular endothelial growth factor.
Figures
Similar articles
-
From Molecular Mechanisms to Clinical Management of Antineoplastic Drug-Induced Cardiovascular Toxicity: A Translational Overview.Antioxid Redox Signal. 2019 Jun 20;30(18):2110-2153. doi: 10.1089/ars.2016.6930. Epub 2017 May 15. Antioxid Redox Signal. 2019. PMID: 28398124 Free PMC article. Review.
-
Oxidative stress in anticancer therapies-related cardiac dysfunction.Free Radic Biol Med. 2021 Jun;169:410-415. doi: 10.1016/j.freeradbiomed.2021.04.021. Epub 2021 Apr 28. Free Radic Biol Med. 2021. PMID: 33930514
-
Redox interactions-induced cardiac toxicity in cancer therapy.Arch Biochem Biophys. 2021 Sep 15;708:108952. doi: 10.1016/j.abb.2021.108952. Epub 2021 Jun 7. Arch Biochem Biophys. 2021. PMID: 34097901 Review.
-
Effects of anticancer drugs on the cardiac mitochondrial toxicity and their underlying mechanisms for novel cardiac protective strategies.Life Sci. 2021 Jul 15;277:119607. doi: 10.1016/j.lfs.2021.119607. Epub 2021 May 13. Life Sci. 2021. PMID: 33992675 Review.
-
Antioxidant Approach as a Cardioprotective Strategy in Chemotherapy-Induced Cardiotoxicity.Antioxid Redox Signal. 2021 Mar 1;34(7):572-588. doi: 10.1089/ars.2020.8055. Epub 2020 Apr 9. Antioxid Redox Signal. 2021. PMID: 32151144 Review.
Cited by
-
Osteoporosis in Childhood Cancer Survivors: Physiopathology, Prevention, Therapy and Future Perspectives.Cancers (Basel). 2022 Sep 6;14(18):4349. doi: 10.3390/cancers14184349. Cancers (Basel). 2022. PMID: 36139510 Free PMC article. Review.
-
New Insights in Early Detection of Anticancer Drug-Related Cardiotoxicity Using Perfusion and Metabolic Imaging.Front Cardiovasc Med. 2022 Feb 7;9:813883. doi: 10.3389/fcvm.2022.813883. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35198613 Free PMC article. Review.
-
Cancer Risk in the Heart Failure Population: Epidemiology, Mechanisms, and Clinical Implications.Curr Oncol Rep. 2020 Dec 2;23(1):7. doi: 10.1007/s11912-020-00990-z. Curr Oncol Rep. 2020. PMID: 33263821 Free PMC article. Review.
-
Involvement of Abnormal Gut Microbiota Composition and Function in Doxorubicin-Induced Cardiotoxicity.Front Cell Infect Microbiol. 2022 Feb 25;12:808837. doi: 10.3389/fcimb.2022.808837. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35281446 Free PMC article.
-
Cardiovascular Risk in Childhood Cancer Survivors.Biomedicines. 2022 Dec 1;10(12):3098. doi: 10.3390/biomedicines10123098. Biomedicines. 2022. PMID: 36551851 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials