

Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity
- PMID: 29556237
- PMCID: PMC5844972
- DOI: 10.3389/fimmu.2018.00422
Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity
Abstract
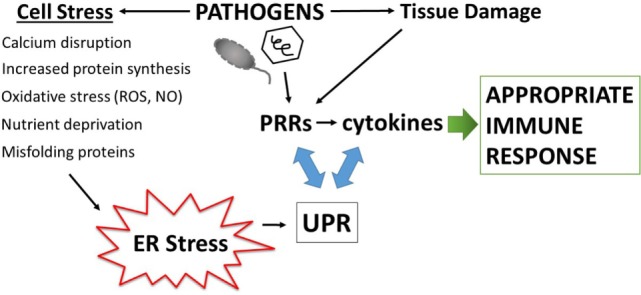
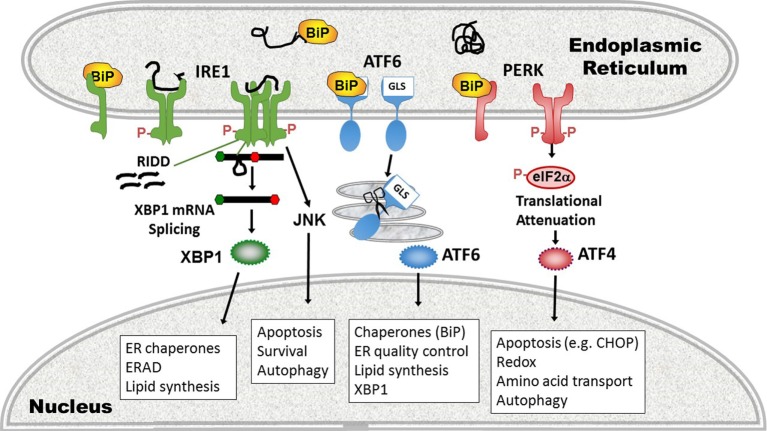
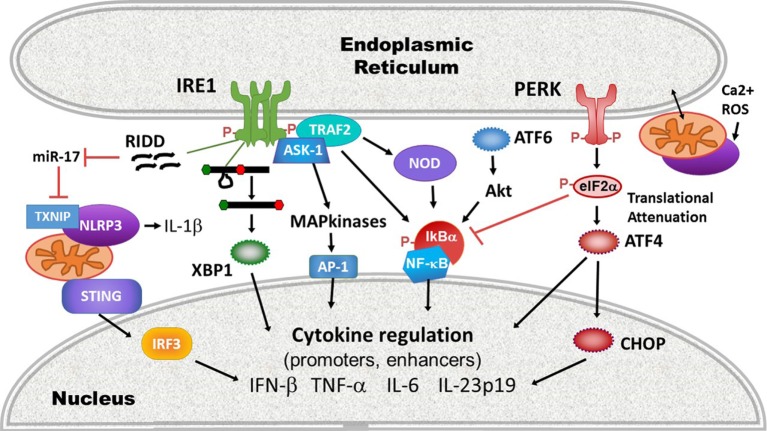
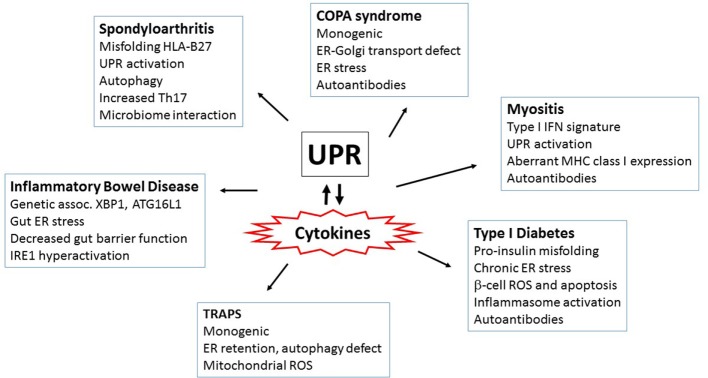
Protein folding in the endoplasmic reticulum (ER) is an essential cell function. To safeguard this process in the face of environmental threats and internal stressors, cells mount an evolutionarily conserved response known as the unfolded protein response (UPR). Invading pathogens induce cellular stress that impacts protein folding, thus the UPR is well situated to sense danger and contribute to immune responses. Cytokines (inflammatory cytokines and interferons) critically mediate host defense against pathogens, but when aberrantly produced, may also drive pathologic inflammation. The UPR influences cytokine production on multiple levels, from stimulation of pattern recognition receptors, to modulation of inflammatory signaling pathways, and the regulation of cytokine transcription factors. This review will focus on the mechanisms underlying cytokine regulation by the UPR, and the repercussions of this relationship for infection and autoimmune/autoinflammatory diseases. Interrogation of viral and bacterial infections has revealed increasing numbers of examples where pathogens induce or modulate the UPR and implicated UPR-modulated cytokines in host response. The flip side of this coin, the UPR/ER stress responses have been increasingly recognized in a variety of autoimmune and inflammatory diseases. Examples include monogenic disorders of ER function, diseases linked to misfolding protein (HLA-B27 and spondyloarthritis), diseases directly implicating UPR and autophagy genes (inflammatory bowel disease), and autoimmune diseases targeting highly secretory cells (e.g., diabetes). Given the burgeoning interest in pharmacologically targeting the UPR, greater discernment is needed regarding how the UPR regulates cytokine production during specific infections and autoimmune processes, and the relative place of this interaction in pathogenesis.
Keywords: autoimmunity; autoinflammatory disease; bacteria; cytokine regulation; endoplasmic reticulum stress; infection; unfolded protein response; virus.
Figures
Similar articles
-
The role of the unfolded protein response in axial spondyloarthritis.Clin Rheumatol. 2016 Jun;35(6):1425-31. doi: 10.1007/s10067-015-3117-5. Epub 2015 Nov 14. Clin Rheumatol. 2016. PMID: 26567900 Review.
-
Unfolded protein responses in the intestinal epithelium: sensors for the microbial and metabolic environment.J Clin Gastroenterol. 2012 Oct;46 Suppl:S3-5. doi: 10.1097/MCG.0b013e318264e632. J Clin Gastroenterol. 2012. PMID: 22955354
-
HLA-B27 misfolding and ankylosing spondylitis.Mol Immunol. 2014 Jan;57(1):44-51. doi: 10.1016/j.molimm.2013.07.013. Epub 2013 Aug 30. Mol Immunol. 2014. PMID: 23993278 Free PMC article. Review.
-
Endoplasmic reticulum stress in autoimmune diseases: Can altered protein quality control and/or unfolded protein response contribute to autoimmunity? A critical review on Sjögren's syndrome.Autoimmun Rev. 2018 Aug;17(8):796-808. doi: 10.1016/j.autrev.2018.02.009. Epub 2018 Jun 8. Autoimmun Rev. 2018. PMID: 29890347 Review.
-
IRE1α Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases.Front Immunol. 2018 Jun 6;9:1289. doi: 10.3389/fimmu.2018.01289. eCollection 2018. Front Immunol. 2018. PMID: 29928282 Free PMC article. Review.
Cited by
-
The Role of TRAIL/DRs in the Modulation of Immune Cells and Responses.Cancers (Basel). 2019 Sep 30;11(10):1469. doi: 10.3390/cancers11101469. Cancers (Basel). 2019. PMID: 31574961 Free PMC article. Review.
-
P53 versus inflammation: an update.Cell Cycle. 2020 Jan;19(2):160-162. doi: 10.1080/15384101.2019.1708575. Epub 2019 Dec 27. Cell Cycle. 2020. PMID: 31880200 Free PMC article. Review.
-
Molecular Insight Into the IRE1α-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain.Front Immunol. 2019 Dec 18;10:2900. doi: 10.3389/fimmu.2019.02900. eCollection 2019. Front Immunol. 2019. PMID: 31921161 Free PMC article.
-
SARS-CoV-2 diverges from other betacoronaviruses in only partially activating the IRE1α/XBP1 ER stress pathway in human lung-derived cells.bioRxiv [Preprint]. 2022 Jun 13:2021.12.30.474519. doi: 10.1101/2021.12.30.474519. bioRxiv. 2022. Update in: mBio. 2022 Oct 26;13(5):e0241522. doi: 10.1128/mbio.02415-22. PMID: 35821981 Free PMC article. Updated. Preprint.
-
Navigating PRKCSH's impact on cancer: from N-linked glycosylation to death pathway and anti-tumor immunity.Front Oncol. 2024 Mar 20;14:1378694. doi: 10.3389/fonc.2024.1378694. eCollection 2024. Front Oncol. 2024. PMID: 38571496 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials