

The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis
- PMID: 29518028
- PMCID: PMC5877639
- DOI: 10.3390/ijms19030778
The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis
Abstract
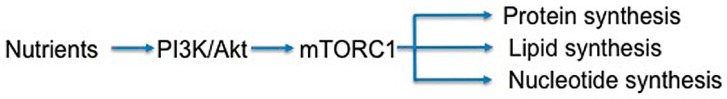
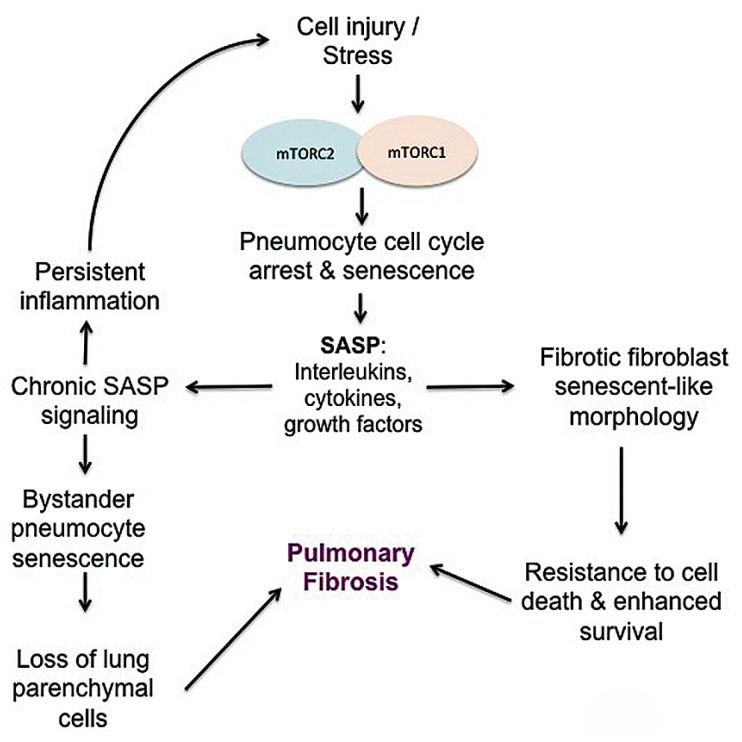
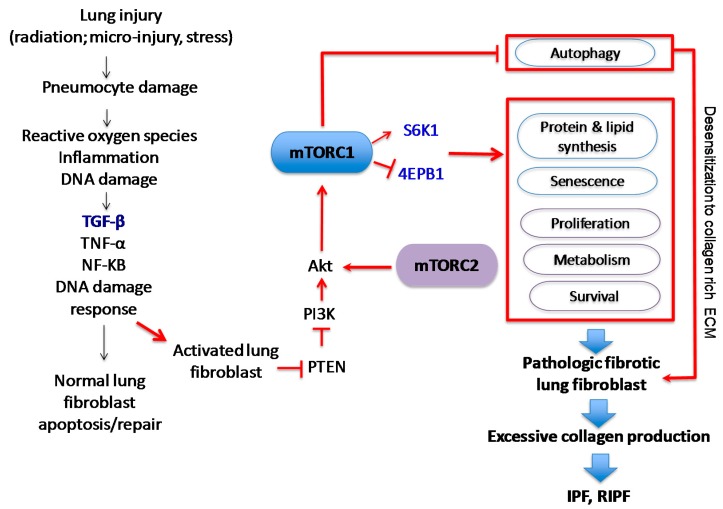
The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR)-dependent pathway is one of the most integral pathways linked to cell metabolism, proliferation, differentiation, and survival. This pathway is dysregulated in a variety of diseases, including neoplasia, immune-mediated diseases, and fibroproliferative diseases such as pulmonary fibrosis. The mTOR kinase is frequently referred to as the master regulator of this pathway. Alterations in mTOR signaling are closely associated with dysregulation of autophagy, inflammation, and cell growth and survival, leading to the development of lung fibrosis. Inhibitors of mTOR have been widely studied in cancer therapy, as they may sensitize cancer cells to radiation therapy. Studies also suggest that mTOR inhibitors are promising modulators of fibroproliferative diseases such as idiopathic pulmonary fibrosis (IPF) and radiation-induced pulmonary fibrosis (RIPF). Therefore, mTOR represents an attractive and unique therapeutic target in pulmonary fibrosis. In this review, we discuss the pathological role of mTOR kinase in pulmonary fibrosis and examine how mTOR inhibitors may mitigate fibrotic progression.
Keywords: fibrosis; idiopathic pulmonary fibrosis (IPF); mammalian target of rapamycin (mTOR); phosphoinositide 3-kinase (PI3K); protein kinase B (AKT); radiation-induced pulmonary fibrosis (RIPF).
Conflict of interest statement
The authors declare no conflict of interest.
Figures




Similar articles
-
Ligustrazin increases lung cell autophagy and ameliorates paraquat-induced pulmonary fibrosis by inhibiting PI3K/Akt/mTOR and hedgehog signalling via increasing miR-193a expression.BMC Pulm Med. 2019 Feb 11;19(1):35. doi: 10.1186/s12890-019-0799-5. BMC Pulm Med. 2019. PMID: 30744607 Free PMC article.
-
The Role of mTOR Signaling as a Therapeutic Target in Cancer.Int J Mol Sci. 2021 Feb 9;22(4):1743. doi: 10.3390/ijms22041743. Int J Mol Sci. 2021. PMID: 33572326 Free PMC article. Review.
-
Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma.Cancer Treat Rev. 2014 Sep;40(8):980-9. doi: 10.1016/j.ctrv.2014.06.006. Epub 2014 Jul 3. Cancer Treat Rev. 2014. PMID: 25037117 Review.
-
Targeting the PI3K/mTOR pathway in idiopathic pulmonary fibrosis: Advances and therapeutic potential.Bioorg Med Chem. 2024 Dec 1;115:117908. doi: 10.1016/j.bmc.2024.117908. Epub 2024 Sep 5. Bioorg Med Chem. 2024. PMID: 39471771 Review.
-
PI3K/Akt signaling is involved in the pathogenesis of bleomycin‑induced pulmonary fibrosis via regulation of epithelial‑mesenchymal transition.Mol Med Rep. 2016 Dec;14(6):5699-5706. doi: 10.3892/mmr.2016.5960. Epub 2016 Nov 22. Mol Med Rep. 2016. PMID: 27878273
Cited by
-
Roles of exosomes and exosome-derived miRNAs in pulmonary fibrosis.Front Pharmacol. 2022 Aug 11;13:928933. doi: 10.3389/fphar.2022.928933. eCollection 2022. Front Pharmacol. 2022. PMID: 36034858 Free PMC article. Review.
-
Hyperactive mTORC1 in lung mesenchyme induces endothelial cell dysfunction and pulmonary vascular remodeling.J Clin Invest. 2023 Dec 20;134(4):e172116. doi: 10.1172/JCI172116. J Clin Invest. 2023. PMID: 38127441 Free PMC article.
-
Update on Novel Targeted Therapy for Pleural Organization and Fibrosis.Int J Mol Sci. 2022 Jan 29;23(3):1587. doi: 10.3390/ijms23031587. Int J Mol Sci. 2022. PMID: 35163509 Free PMC article. Review.
-
The Akt pathway in oncology therapy and beyond (Review).Int J Oncol. 2018 Dec;53(6):2319-2331. doi: 10.3892/ijo.2018.4597. Epub 2018 Oct 16. Int J Oncol. 2018. PMID: 30334567 Free PMC article. Review.
-
The changing landscape of thyroid eye disease: current clinical advances and future outlook.Eye (Lond). 2024 Jun;38(8):1425-1437. doi: 10.1038/s41433-024-02967-9. Epub 2024 Feb 19. Eye (Lond). 2024. PMID: 38374366 Free PMC article. Review.
References
-
- Rosenbloom J., Macarak E., Piera-Velazquez S., Jimenez S.A. Human fibrotic diseases: Current challenges in fibrosis research. Methods Mol. Biol. 2017;1627:1–23. - PubMed
-
- Calio A., Lever V., Rossi A., Gilioli E., Brunelli M., Dubini A., Tomassetti S., Piciucchi S., Nottegar A., Rossi G., et al. Increased frequency of bronchiolar histotypes in lung carcinomas associated with idiopathic pulmonary fibrosis. Histopathology. 2017;71:725–735. doi: 10.1111/his.13269. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
