

Hypertrophic cardiomyopathy mutation R58Q in the myosin regulatory light chain perturbs thick filament-based regulation in cardiac muscle
- PMID: 29452157
- PMCID: PMC5883317
- DOI: 10.1016/j.yjmcc.2018.02.009
Hypertrophic cardiomyopathy mutation R58Q in the myosin regulatory light chain perturbs thick filament-based regulation in cardiac muscle
Abstract
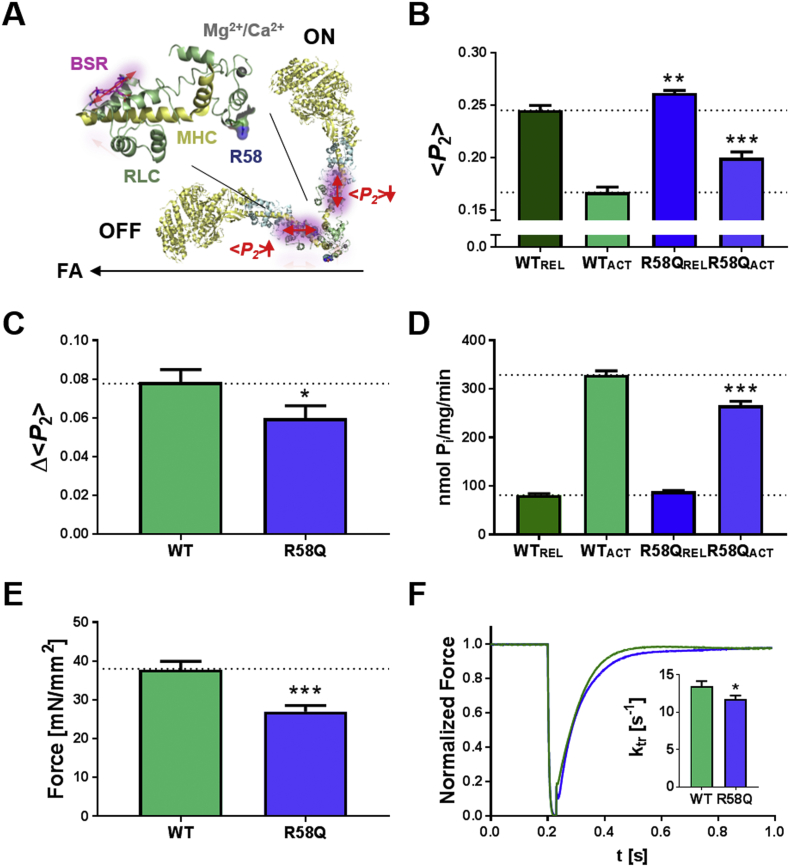
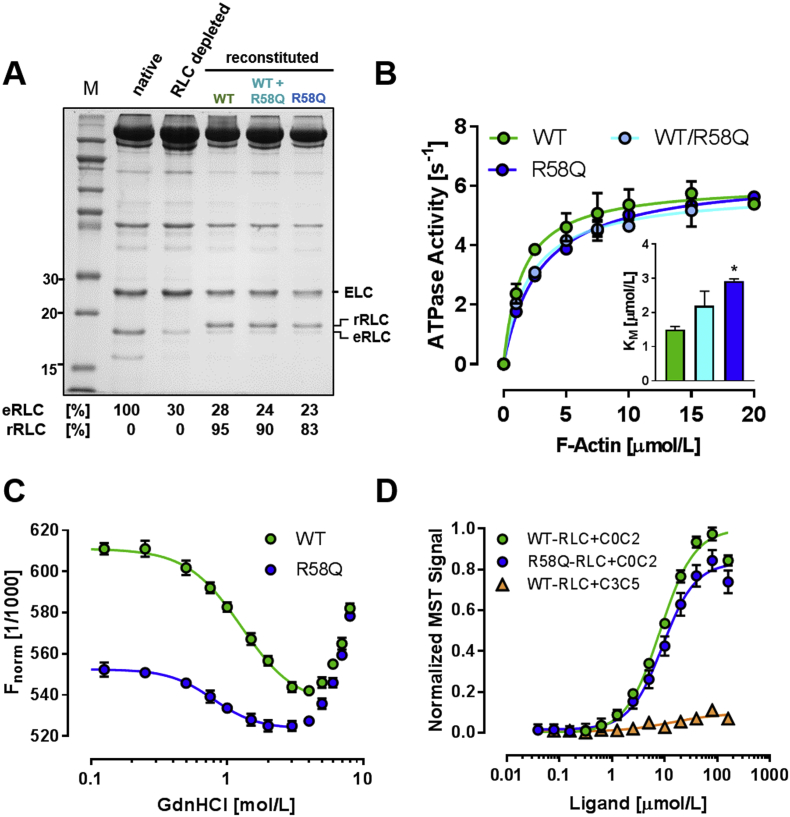
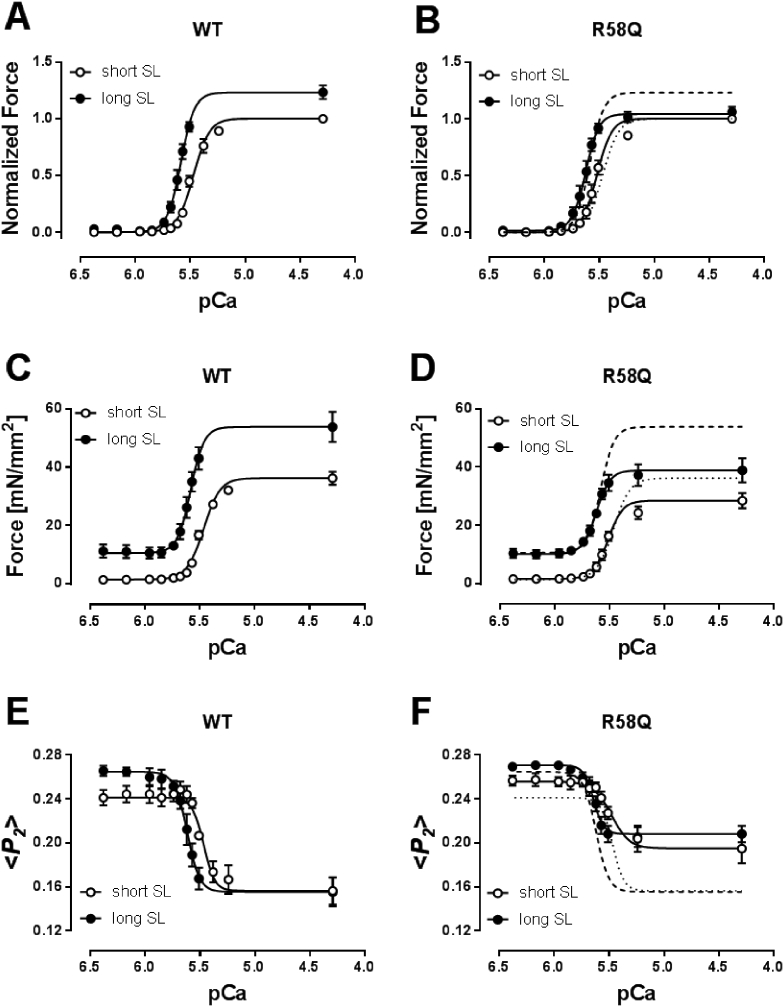
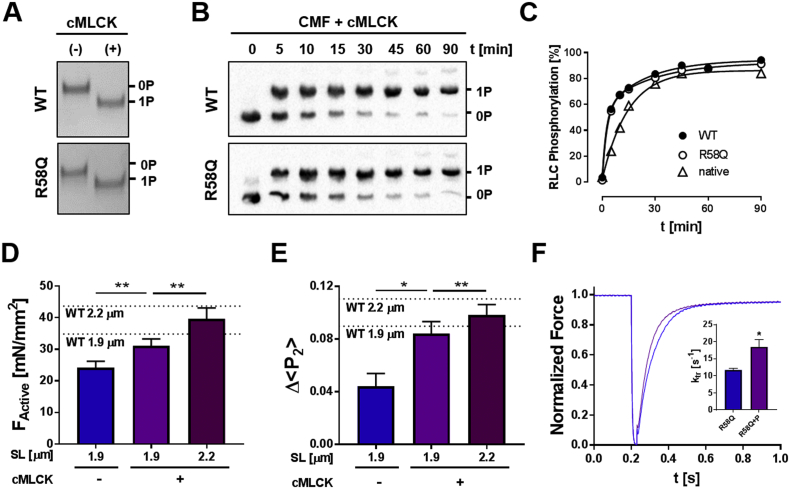
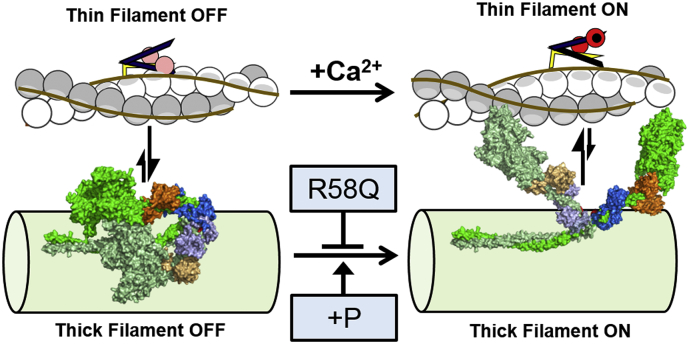
Hypertrophic cardiomyopathy (HCM) is frequently linked to mutations in the protein components of the myosin-containing thick filaments leading to contractile dysfunction and ultimately heart failure. However, the molecular structure-function relationships that underlie these pathological effects remain largely obscure. Here we chose an example mutation (R58Q) in the myosin regulatory light chain (RLC) that is associated with a severe HCM phenotype and combined the results from a wide range of in vitro and in situ structural and functional studies on isolated protein components, myofibrils and ventricular trabeculae to create an extensive map of structure-function relationships. The results can be understood in terms of a unifying hypothesis that illuminates both the effects of the mutation and physiological signaling pathways. R58Q promotes an OFF state of the thick filaments that reduces the number of myosin head domains that are available for actin interaction and ATP utilization. Moreover this mutation uncouples two aspects of length-dependent activation (LDA), the cellular basis of the Frank-Starling relation that couples cardiac output to venous return; R58Q reduces maximum calcium-activated force with no significant effect on myofilament calcium sensitivity. Finally, phosphorylation of R58Q-RLC to levels that may be relevant both physiologically and pathologically restores the regulatory state of the thick filament and the effect of sarcomere length on maximum calcium-activated force and thick filament structure, as well as increasing calcium sensitivity. We conclude that perturbation of thick filament-based regulation may be a common mechanism in the etiology of missense mutation-associated HCM, and that this signaling pathway offers a promising target for the development of novel therapeutics.
Keywords: Cardiac muscle regulation; Hypertrophic cardiomyopathy; Myosin; Myosin regulatory light chain; Polarized fluorescence.
Copyright © 2018 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
Similar articles
-
Phosphomimetic-mediated in vitro rescue of hypertrophic cardiomyopathy linked to R58Q mutation in myosin regulatory light chain.FEBS J. 2019 Jan;286(1):151-168. doi: 10.1111/febs.14702. Epub 2018 Dec 1. FEBS J. 2019. PMID: 30430732 Free PMC article.
-
Slow-twitch skeletal muscle defects accompany cardiac dysfunction in transgenic mice with a mutation in the myosin regulatory light chain.FASEB J. 2019 Mar;33(3):3152-3166. doi: 10.1096/fj.201801402R. Epub 2018 Oct 26. FASEB J. 2019. PMID: 30365366 Free PMC article.
-
Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice.Cardiovasc Res. 2009 Apr 1;82(1):84-92. doi: 10.1093/cvr/cvp016. Epub 2009 Jan 15. Cardiovasc Res. 2009. PMID: 19150977 Free PMC article.
-
Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations.J Muscle Res Cell Motil. 2015 Dec;36(6):433-45. doi: 10.1007/s10974-015-9423-3. Epub 2015 Sep 18. J Muscle Res Cell Motil. 2015. PMID: 26385864 Free PMC article. Review.
-
In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament.Circ Res. 2011 Mar 18;108(6):751-64. doi: 10.1161/CIRCRESAHA.110.231670. Circ Res. 2011. PMID: 21415409 Free PMC article. Review.
Cited by
-
Phosphomimetic-mediated in vitro rescue of hypertrophic cardiomyopathy linked to R58Q mutation in myosin regulatory light chain.FEBS J. 2019 Jan;286(1):151-168. doi: 10.1111/febs.14702. Epub 2018 Dec 1. FEBS J. 2019. PMID: 30430732 Free PMC article.
-
Ablation of the N terminus of cardiac essential light chain promotes the super-relaxed state of myosin and counteracts hypercontractility in hypertrophic cardiomyopathy mutant mice.FEBS J. 2020 Sep;287(18):3989-4004. doi: 10.1111/febs.15243. Epub 2020 Feb 25. FEBS J. 2020. PMID: 32034976 Free PMC article.
-
Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin.Proc Natl Acad Sci U S A. 2018 Aug 7;115(32):E7486-E7494. doi: 10.1073/pnas.1720342115. Epub 2018 Jul 17. Proc Natl Acad Sci U S A. 2018. PMID: 30018063 Free PMC article.
-
Slow-twitch skeletal muscle defects accompany cardiac dysfunction in transgenic mice with a mutation in the myosin regulatory light chain.FASEB J. 2019 Mar;33(3):3152-3166. doi: 10.1096/fj.201801402R. Epub 2018 Oct 26. FASEB J. 2019. PMID: 30365366 Free PMC article.
-
Proposed mechanism for the length dependence of the force developed in maximally activated muscles.Sci Rep. 2019 Feb 4;9(1):1317. doi: 10.1038/s41598-018-36706-4. Sci Rep. 2019. PMID: 30718530 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
