

Methods for CpG Methylation Array Profiling Via Bisulfite Conversion
- PMID: 29423802
- PMCID: PMC6548194
- DOI: 10.1007/978-1-4939-7471-9_13
Methods for CpG Methylation Array Profiling Via Bisulfite Conversion
Abstract
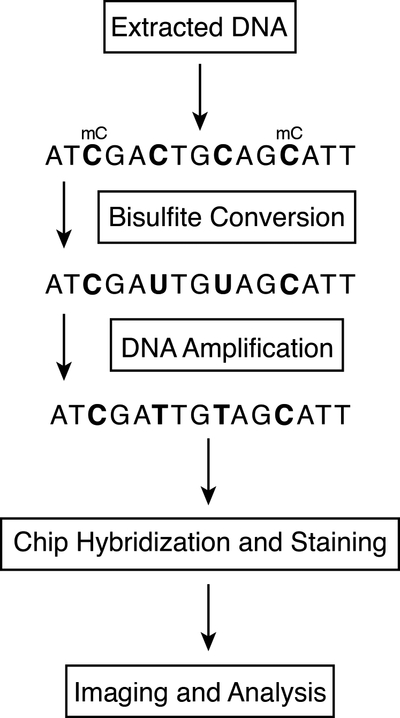
DNA methylation is a key factor in epigenetic regulation, and contributes to the pathogenesis of many diseases, including various forms of cancers, and epigenetic events such X inactivation, cellular differentiation and proliferation, and embryonic development. The most conserved epigenetic modification in plants, animals, and fungi is 5-methylcytosine (5mC), which has been well characterized across a diverse range of species. Many technologies have been developed to measure modifications in methylation with respect to biological processes, and the most common method, long considered a gold standard for identifying regions of methylation, is bisulfite conversion. In this technique, DNA is treated with bisulfite, which converts cytosine residues to uracil, but does not affect cytosine residues that have been methylated, such as 5-methylcytosines. Following bisulfite conversion, the only cytosine residues remaining in the DNA, therefore, are those that have been methylated. Subsequent sequencing can then distinguish between unmethylated cytosines, which are displayed as thymines in the resulting amplified sequence of the sense strand, and 5-methylcytosines, which are displayed as cytosines in the resulting amplified sequence of the sense strand, at the single nucleotide level. In this chapter, we describe an array-based protocol for identifying methylated DNA regions. We discuss protocols for DNA quantification, bisulfite conversion, library preparation, and chip assembly, and present an overview of current methods for the analysis of methylation data.
Keywords: Bisulfite conversion; CpG; DNA; Epigenetics; Methylation.
Figures
Similar articles
-
Investigation of genomic methylation status using methylation-specific and bisulfite sequencing polymerase chain reaction.Methods Mol Biol. 2015;1288:193-212. doi: 10.1007/978-1-4939-2474-5_11. Methods Mol Biol. 2015. PMID: 25827881
-
Monitoring methylation changes in cancer.Adv Biochem Eng Biotechnol. 2007;104:1-11. doi: 10.1007/10_024. Adv Biochem Eng Biotechnol. 2007. PMID: 17290816 Review.
-
High-speed conversion of cytosine to uracil in bisulfite genomic sequencing analysis of DNA methylation.DNA Res. 2004 Dec 31;11(6):409-15. doi: 10.1093/dnares/11.6.409. DNA Res. 2004. PMID: 15871463
-
Cytosines adjacent to methylated CpG sites can be partially resistant to conversion in genomic bisulfite sequencing leading to methylation artifacts.Anal Biochem. 1998 Nov 1;264(1):129-32. doi: 10.1006/abio.1998.2833. Anal Biochem. 1998. PMID: 9784198 No abstract available.
-
Methodological aspects of whole-genome bisulfite sequencing analysis.Brief Bioinform. 2015 May;16(3):369-79. doi: 10.1093/bib/bbu016. Epub 2014 May 27. Brief Bioinform. 2015. PMID: 24867940 Review.
Cited by
-
Coding and Non-Coding Transcriptomic Landscape of Aortic Complications in Marfan Syndrome.Int J Mol Sci. 2024 Jul 5;25(13):7367. doi: 10.3390/ijms25137367. Int J Mol Sci. 2024. PMID: 39000474 Free PMC article. Review.
-
Methods in DNA methylation array dataset analysis: A review.Comput Struct Biotechnol J. 2024 May 17;23:2304-2325. doi: 10.1016/j.csbj.2024.05.015. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 38845821 Free PMC article. Review.
-
Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons.Epigenetics. 2022 May;17(5):518-530. doi: 10.1080/15592294.2021.1924970. Epub 2021 May 11. Epigenetics. 2022. PMID: 33975521 Free PMC article.
-
The DNA demethylation-regulated SFRP2 dictates the progression of endometriosis via activation of the Wnt/β-catenin signaling pathway.BMC Mol Cell Biol. 2023 Mar 29;24(1):12. doi: 10.1186/s12860-023-00470-9. BMC Mol Cell Biol. 2023. PMID: 36991319 Free PMC article.
-
Little to Give, Much to Gain-What Can You Do With a Dried Blood Spot?Curr Environ Health Rep. 2020 Sep;7(3):211-221. doi: 10.1007/s40572-020-00289-y. Curr Environ Health Rep. 2020. PMID: 32851603 Free PMC article. Review.
References
-
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 1985;40(1):91–9. - PubMed
-
- Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187(4173):226–32. - PubMed
-
- Dayeh T, Volkov P, Salo S, Hall E, Nilsson E, Olsson AH, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10(3):e1004160. doi: 10.1371/journal.pgen.1004160. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
