

Reconstructing spatial organizations of chromosomes through manifold learning
- PMID: 29408992
- PMCID: PMC5934626
- DOI: 10.1093/nar/gky065
Reconstructing spatial organizations of chromosomes through manifold learning
Abstract
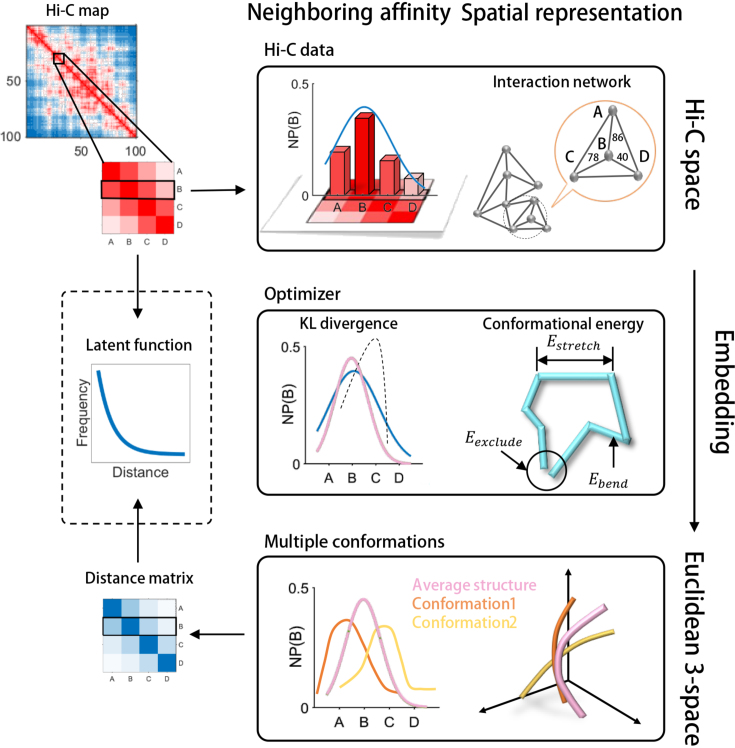
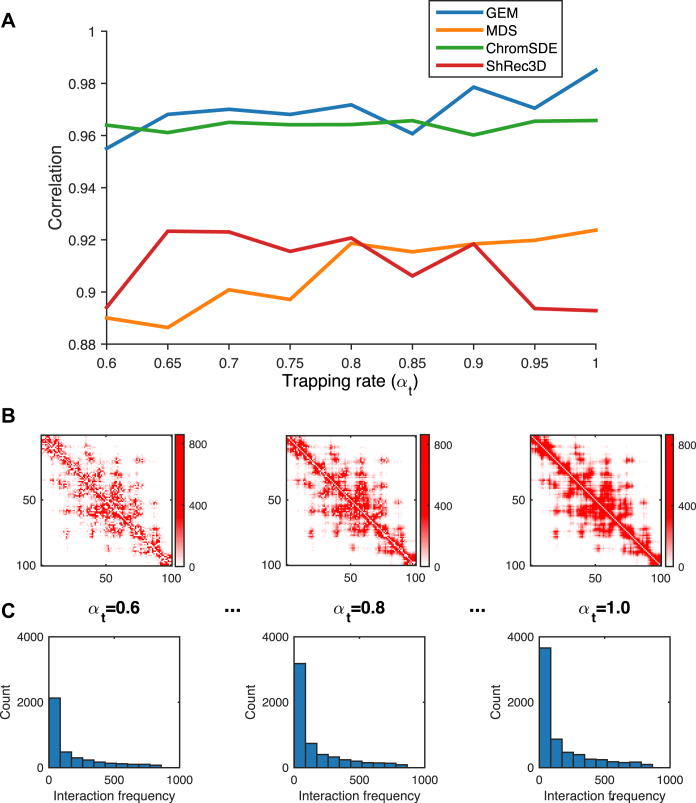
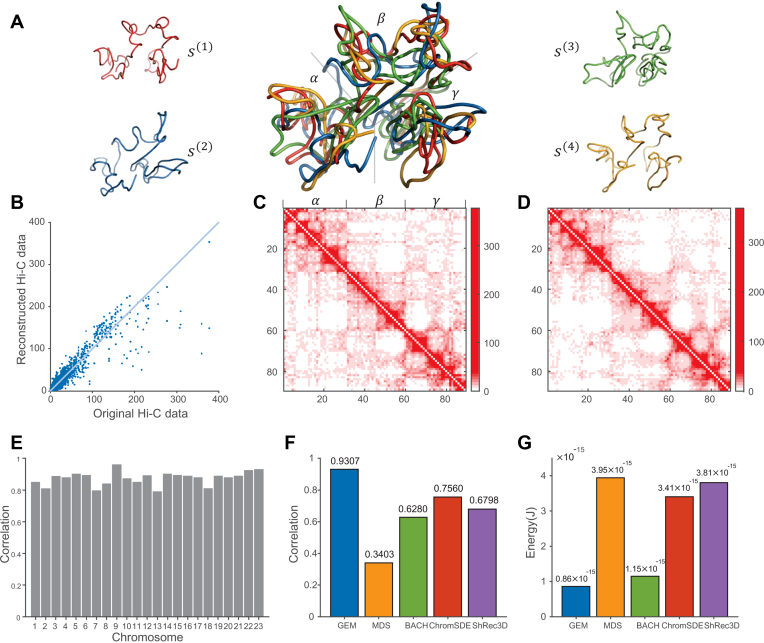
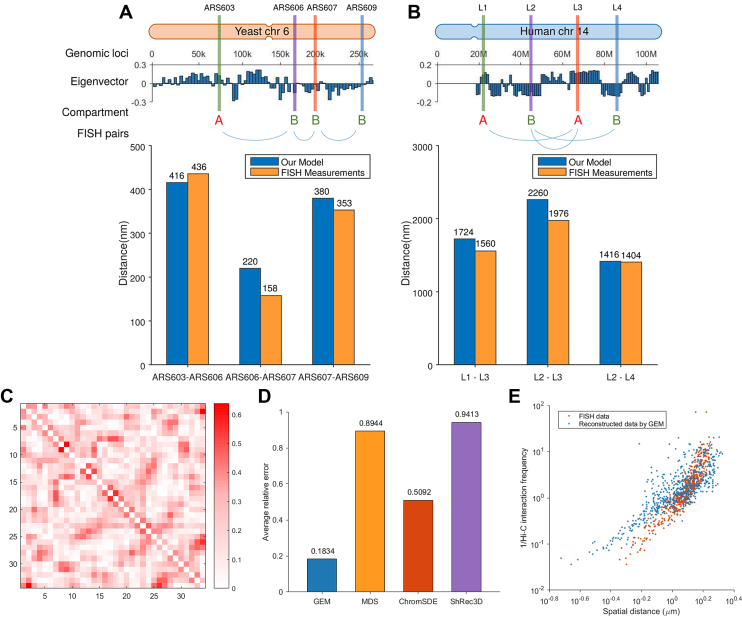
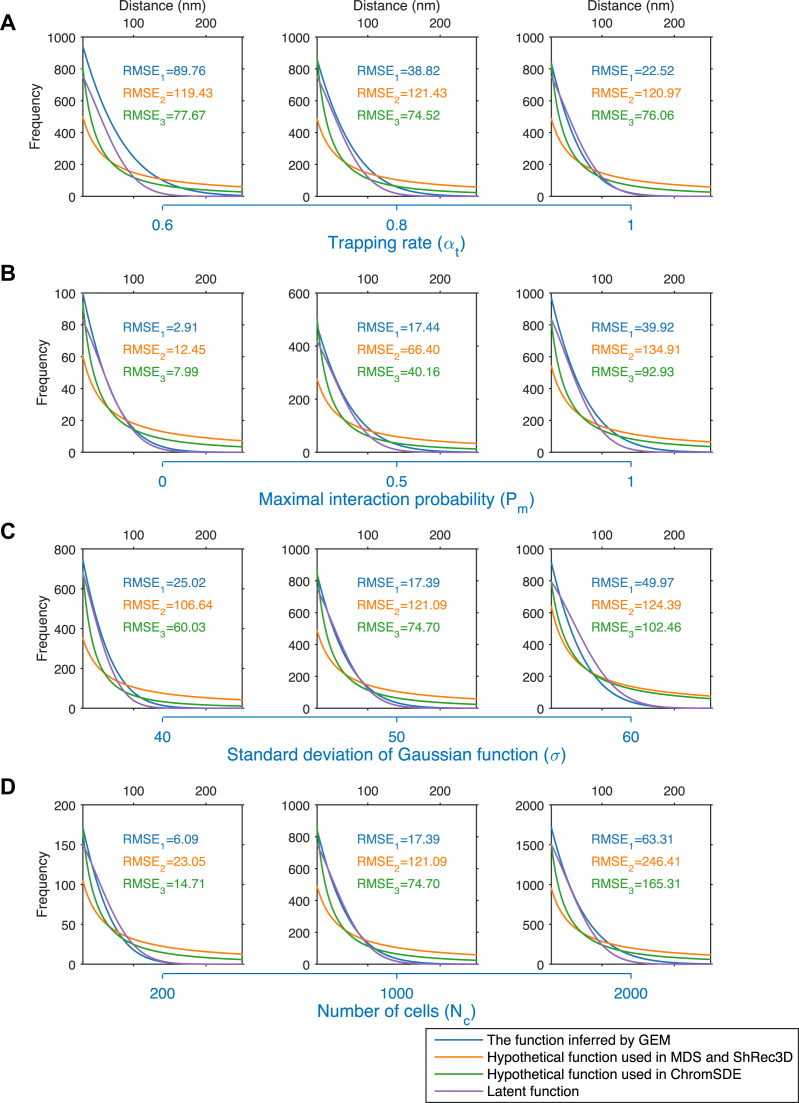
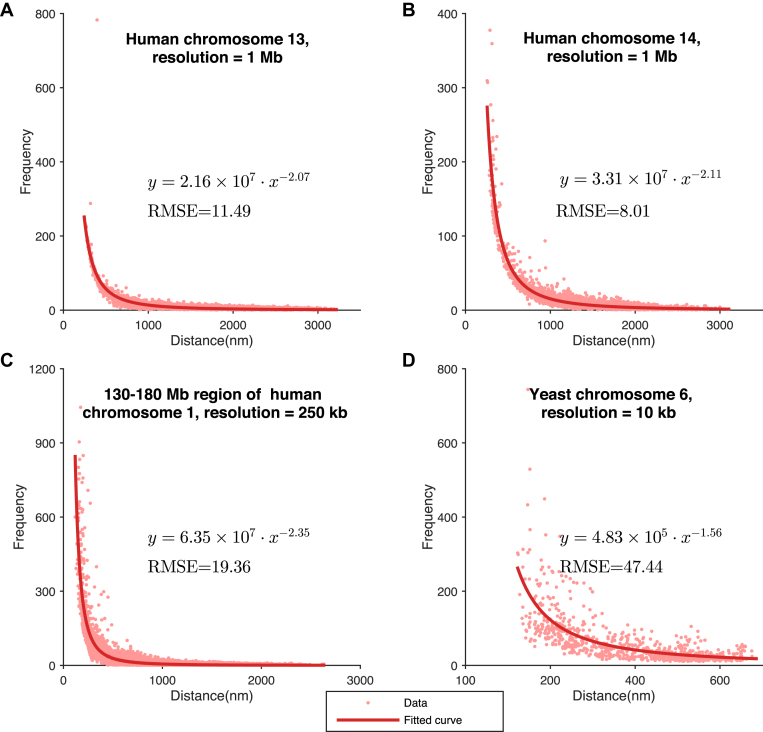
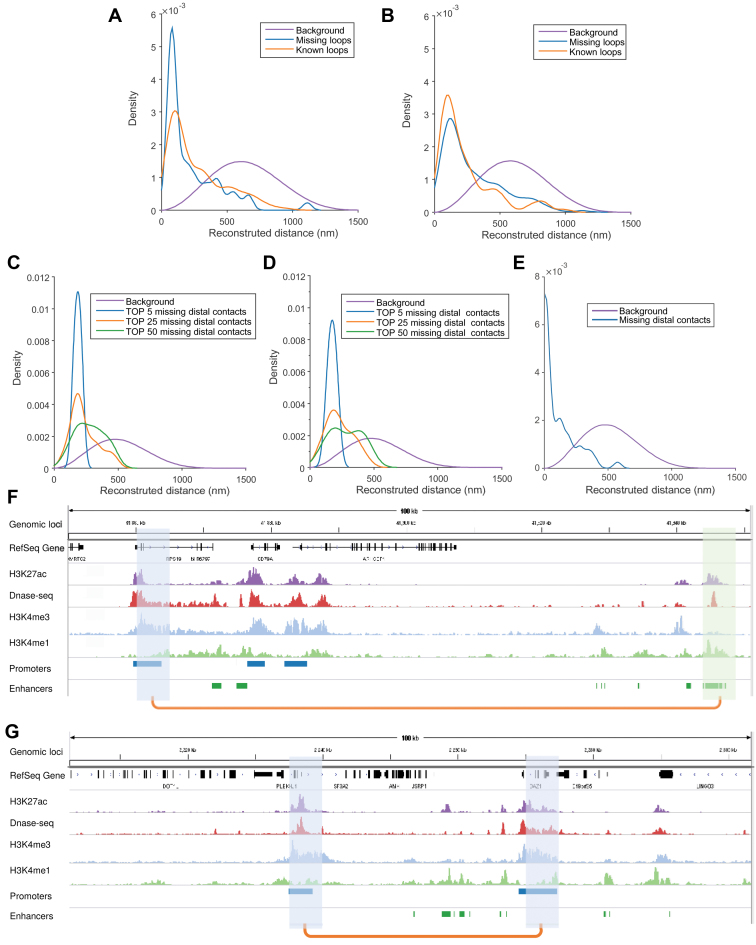
Decoding the spatial organizations of chromosomes has crucial implications for studying eukaryotic gene regulation. Recently, chromosomal conformation capture based technologies, such as Hi-C, have been widely used to uncover the interaction frequencies of genomic loci in a high-throughput and genome-wide manner and provide new insights into the folding of three-dimensional (3D) genome structure. In this paper, we develop a novel manifold learning based framework, called GEM (Genomic organization reconstructor based on conformational Energy and Manifold learning), to reconstruct the three-dimensional organizations of chromosomes by integrating Hi-C data with biophysical feasibility. Unlike previous methods, which explicitly assume specific relationships between Hi-C interaction frequencies and spatial distances, our model directly embeds the neighboring affinities from Hi-C space into 3D Euclidean space. Extensive validations demonstrated that GEM not only greatly outperformed other state-of-art modeling methods but also provided a physically and physiologically valid 3D representations of the organizations of chromosomes. Furthermore, we for the first time apply the modeled chromatin structures to recover long-range genomic interactions missing from original Hi-C data.
Figures
Similar articles
-
A maximum likelihood algorithm for reconstructing 3D structures of human chromosomes from chromosomal contact data.BMC Genomics. 2018 Feb 23;19(1):161. doi: 10.1186/s12864-018-4546-8. BMC Genomics. 2018. PMID: 29471801 Free PMC article.
-
3D genome structure modeling by Lorentzian objective function.Nucleic Acids Res. 2017 Feb 17;45(3):1049-1058. doi: 10.1093/nar/gkw1155. Nucleic Acids Res. 2017. PMID: 28180292 Free PMC article.
-
Integrating Hi-C and FISH data for modeling of the 3D organization of chromosomes.Nat Commun. 2019 May 3;10(1):2049. doi: 10.1038/s41467-019-10005-6. Nat Commun. 2019. PMID: 31053705 Free PMC article.
-
Simulation of different three-dimensional polymer models of interphase chromosomes compared to experiments-an evaluation and review framework of the 3D genome organization.Semin Cell Dev Biol. 2019 Jun;90:19-42. doi: 10.1016/j.semcdb.2018.07.012. Epub 2018 Aug 24. Semin Cell Dev Biol. 2019. PMID: 30125668 Review.
-
Developing novel methods to image and visualize 3D genomes.Cell Biol Toxicol. 2018 Oct;34(5):367-380. doi: 10.1007/s10565-018-9427-z. Epub 2018 Mar 26. Cell Biol Toxicol. 2018. PMID: 29577183 Free PMC article. Review.
Cited by
-
Complementing Hi-C information for 3D chromatin reconstruction by ChromStruct.Front Bioinform. 2024 Jan 22;3:1287168. doi: 10.3389/fbinf.2023.1287168. eCollection 2023. Front Bioinform. 2024. PMID: 38318534 Free PMC article.
-
Chromosome structure modeling tools and their evaluation in bacteria.Brief Bioinform. 2024 Jan 22;25(2):bbae044. doi: 10.1093/bib/bbae044. Brief Bioinform. 2024. PMID: 38385874 Free PMC article. Review.
-
Advances in technologies for 3D genomics research.Sci China Life Sci. 2020 Jun;63(6):811-824. doi: 10.1007/s11427-019-1704-2. Epub 2020 May 8. Sci China Life Sci. 2020. PMID: 32394244 Review.
-
GSDB: a database of 3D chromosome and genome structures reconstructed from Hi-C data.BMC Mol Cell Biol. 2020 Aug 5;21(1):60. doi: 10.1186/s12860-020-00304-y. BMC Mol Cell Biol. 2020. PMID: 32758136 Free PMC article.
-
Subtle changes in chromatin loop contact propensity are associated with differential gene regulation and expression.Nat Commun. 2019 Mar 5;10(1):1054. doi: 10.1038/s41467-019-08940-5. Nat Commun. 2019. PMID: 30837461 Free PMC article.
References
-
- de Laat W., Grosveld F.. Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003; 11:447–459. - PubMed
-
- Fraser P., Bickmore W.. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007; 447:413–417. - PubMed
-
- Cremer T., Cremer C.. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001; 2:292–301. - PubMed
-
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007; 128:787–800. - PubMed
-
- Dekker J., Rippe K., Dekker M., Kleckner N.. Capturing chromosome conformation. Science. 2002; 295:1306–1311. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
