

Sequence analysis of European maize inbred line F2 provides new insights into molecular and chromosomal characteristics of presence/absence variants
- PMID: 29402214
- PMCID: PMC5800051
- DOI: 10.1186/s12864-018-4490-7
Sequence analysis of European maize inbred line F2 provides new insights into molecular and chromosomal characteristics of presence/absence variants
Abstract
Background: Maize is well known for its exceptional structural diversity, including copy number variants (CNVs) and presence/absence variants (PAVs), and there is growing evidence for the role of structural variation in maize adaptation. While PAVs have been described in this important crop species, they have been only scarcely characterized at the sequence level and the extent of presence/absence variation and relative chromosomal landscape of inbred-specific regions remain to be elucidated.
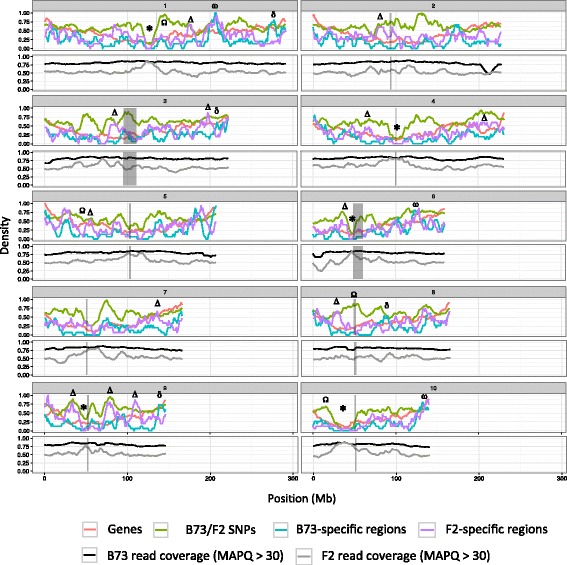
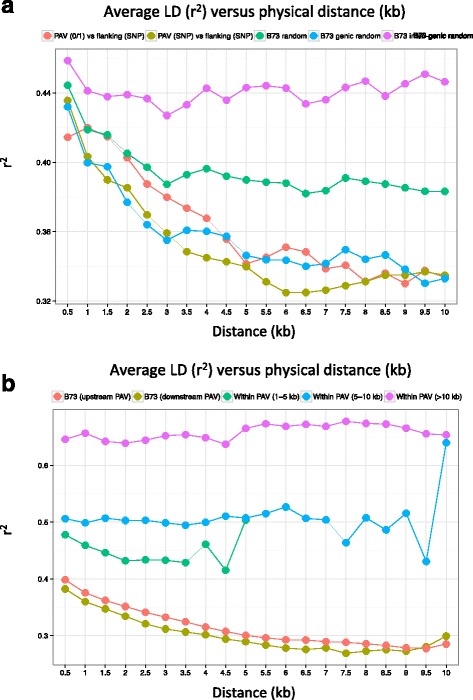
Results: De novo genome sequencing of the French F2 maize inbred line revealed 10,044 novel genomic regions larger than 1 kb, making up 88 Mb of DNA, that are present in F2 but not in B73 (PAV). This set of maize PAV sequences allowed us to annotate PAV content and to analyze sequence breakpoints. Using PAV genotyping on a collection of 25 temperate lines, we also analyzed Linkage Disequilibrium in PAVs and flanking regions, and PAV frequencies within maize genetic groups.
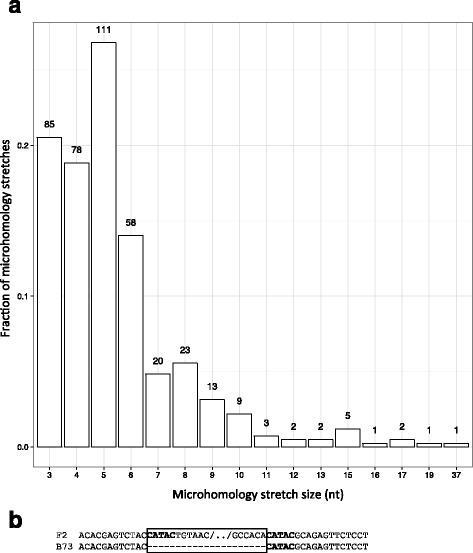
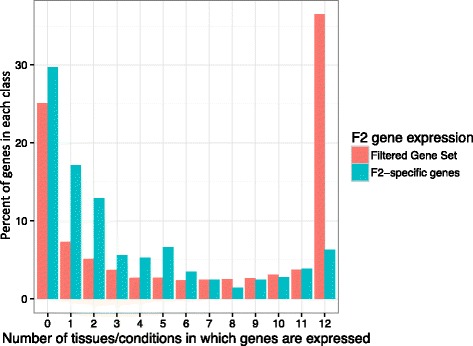
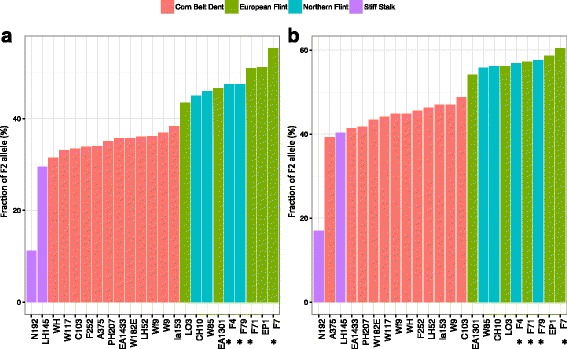
Conclusions: We highlight the possible role of MMEJ-type double strand break repair in maize PAV formation and discover 395 new genes with transcriptional support. Pattern of linkage disequilibrium within PAVs strikingly differs from this of flanking regions and is in accordance with the intuition that PAVs may recombine less than other genomic regions. We show that most PAVs are ancient, while some are found only in European Flint material, thus pinpointing structural features that may be at the origin of adaptive traits involved in the success of this material. Characterization of such PAVs will provide useful material for further association genetic studies in European and temperate maize.
Keywords: De novo assembly; Double strand break repair (DSBR); European germplasm; Genetic diversity; Linkage disequilibrium; Maize; Microhomology mediated end joining (MMEJ); Pan-genome; Presence absence variation (PAV); Structural variation.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content.PLoS Genet. 2009 Nov;5(11):e1000734. doi: 10.1371/journal.pgen.1000734. Epub 2009 Nov 20. PLoS Genet. 2009. PMID: 19956538 Free PMC article.
-
Megabase-scale presence-absence variation with Tripsacum origin was under selection during maize domestication and adaptation.Genome Biol. 2021 Aug 20;22(1):237. doi: 10.1186/s13059-021-02448-2. Genome Biol. 2021. PMID: 34416918 Free PMC article.
-
Genome-wide patterns of large-size presence/absence variants in sorghum.J Integr Plant Biol. 2014 Jan;56(1):24-37. doi: 10.1111/jipb.12121. J Integr Plant Biol. 2014. PMID: 24428208
-
Diversity in global maize germplasm: characterization and utilization.J Biosci. 2012 Nov;37(5):843-55. doi: 10.1007/s12038-012-9227-1. J Biosci. 2012. PMID: 23107920 Review.
-
275 years of forestry meets genomics in Pinus sylvestris.Evol Appl. 2019 Jun 28;13(1):11-30. doi: 10.1111/eva.12809. eCollection 2020 Jan. Evol Appl. 2019. PMID: 31988655 Free PMC article. Review.
Cited by
-
Functional Study of Lipoxygenase-Mediated Resistance against Fusarium verticillioides and Aspergillus flavus Infection in Maize.Int J Mol Sci. 2022 Sep 17;23(18):10894. doi: 10.3390/ijms231810894. Int J Mol Sci. 2022. PMID: 36142806 Free PMC article.
-
Connecting genome structural variation with complex traits in crop plants.Theor Appl Genet. 2019 Mar;132(3):733-750. doi: 10.1007/s00122-018-3233-0. Epub 2018 Nov 17. Theor Appl Genet. 2019. PMID: 30448864 Review.
-
Stability of DNA methylation and chromatin accessibility in structurally diverse maize genomes.G3 (Bethesda). 2021 Aug 7;11(8):jkab190. doi: 10.1093/g3journal/jkab190. G3 (Bethesda). 2021. PMID: 34849810 Free PMC article.
-
Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus.Sci Rep. 2020 Mar 5;10(1):4131. doi: 10.1038/s41598-020-61228-3. Sci Rep. 2020. PMID: 32139810 Free PMC article.
-
Next-Generation Breeding Strategies for Climate-Ready Crops.Front Plant Sci. 2021 Jul 21;12:620420. doi: 10.3389/fpls.2021.620420. eCollection 2021. Front Plant Sci. 2021. PMID: 34367194 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
