

Facilitated Unbinding via Multivalency-Enabled Ternary Complexes: New Paradigm for Protein-DNA Interactions
- PMID: 29368512
- PMCID: PMC5904000
- DOI: 10.1021/acs.accounts.7b00541
Facilitated Unbinding via Multivalency-Enabled Ternary Complexes: New Paradigm for Protein-DNA Interactions
Abstract
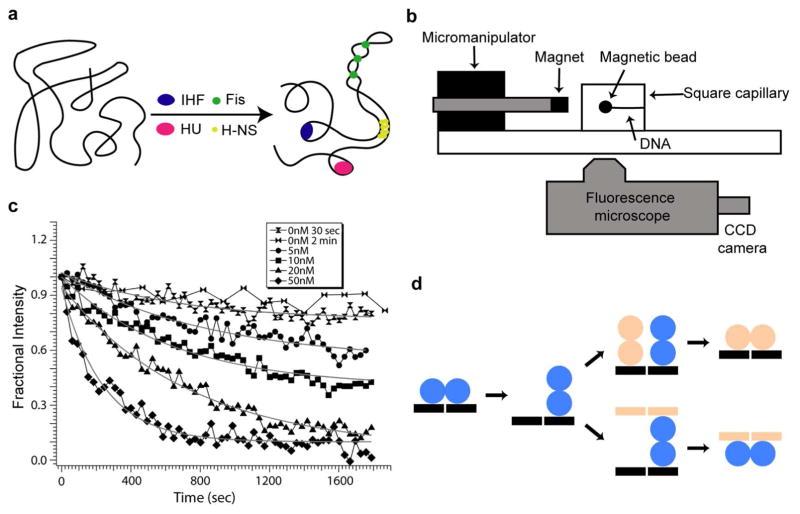
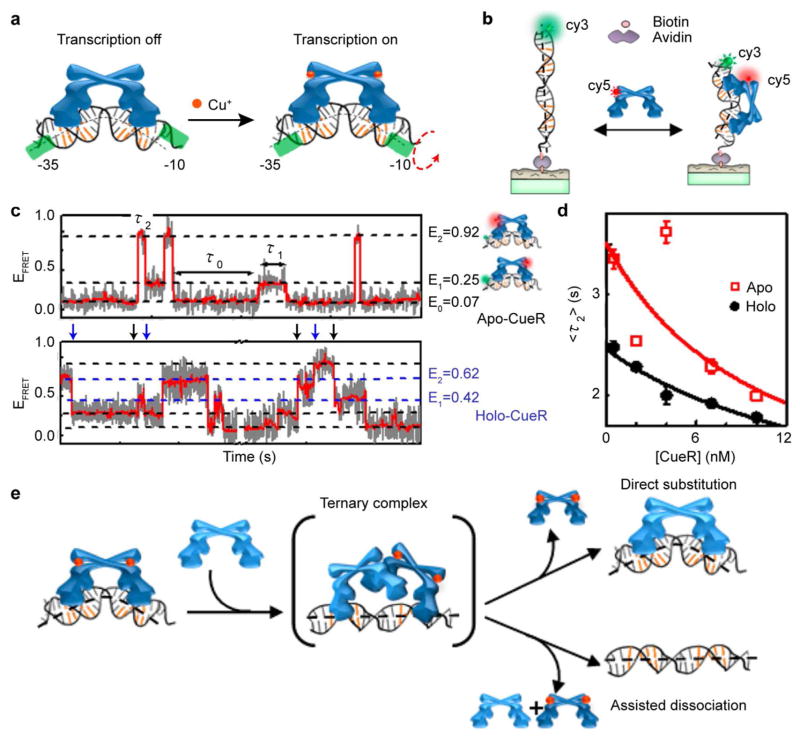
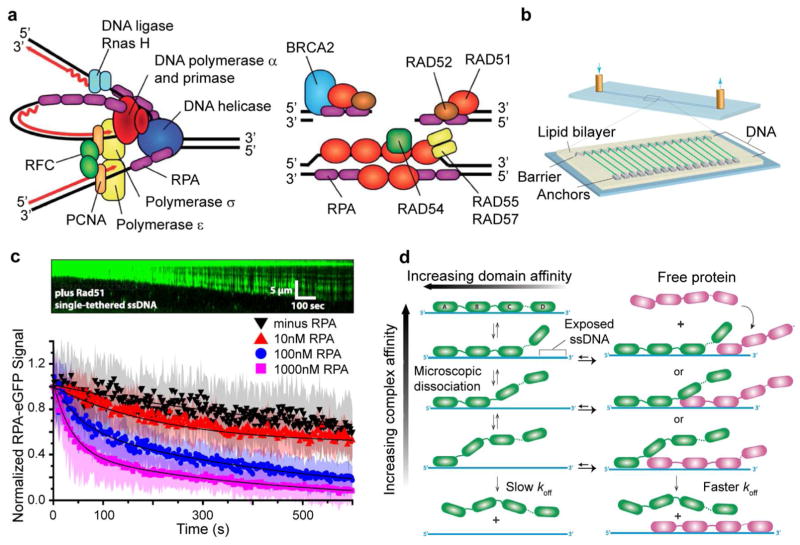
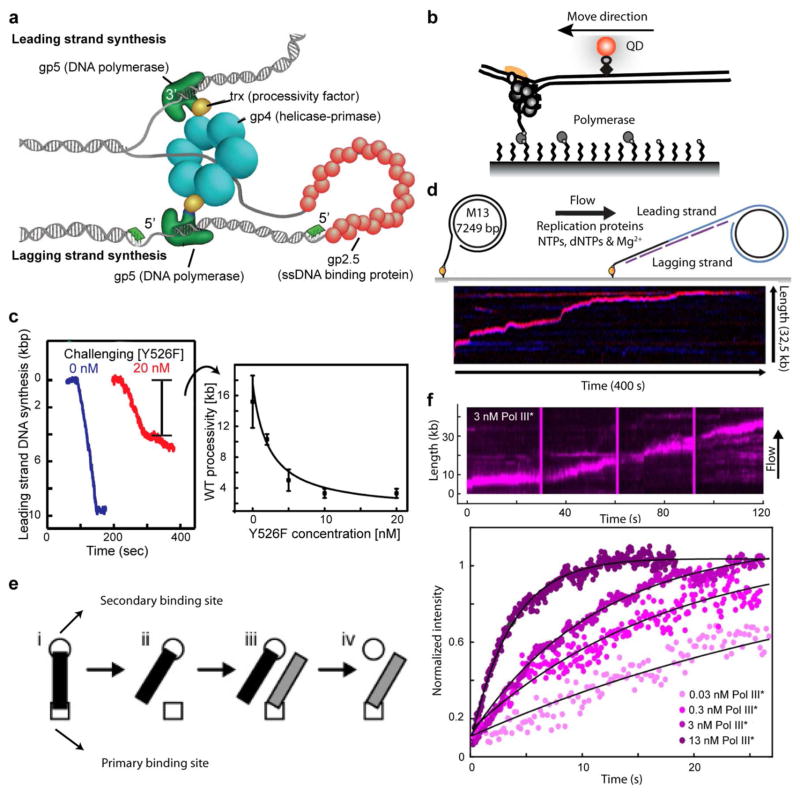
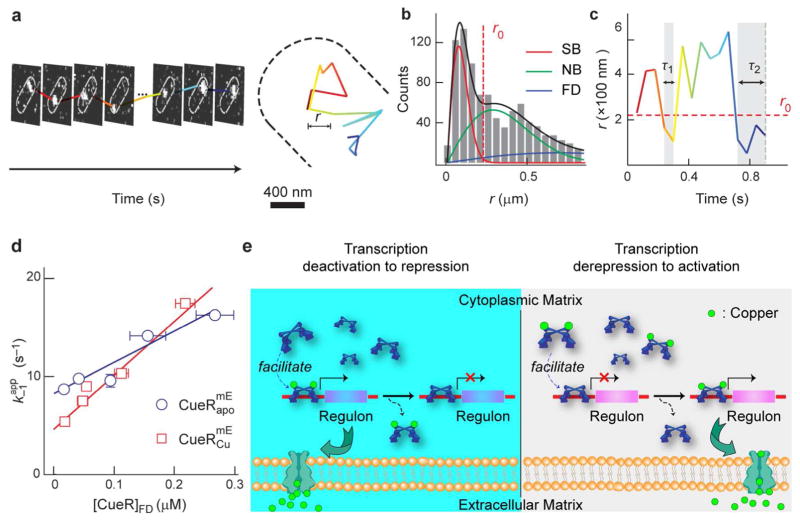
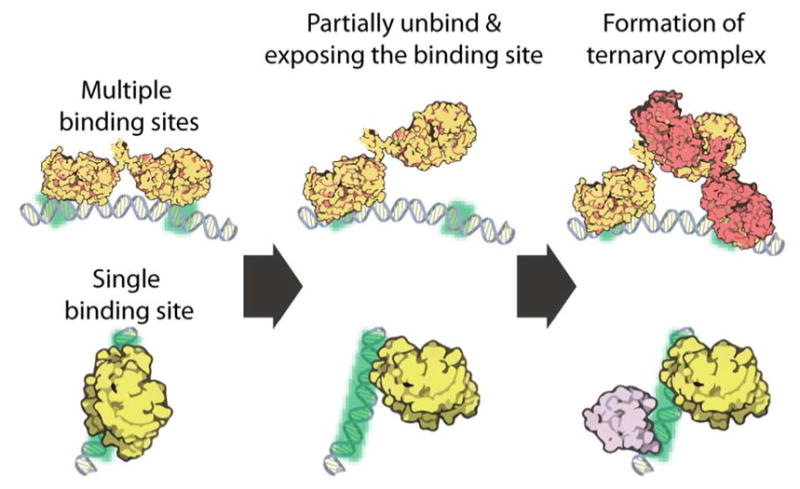
Dynamic protein-DNA interactions constitute highly robust cellular machineries to fulfill cellular functions. A vast number of studies have focused on how DNA-binding proteins search for and interact with their target DNA segments and on what cellular cues can regulate protein binding, for which protein concentration is a most obvious one. In contrast, how protein unbinding could be regulated by protein concentration has evaded attention because protein unbinding from DNA is typically a unimolecular reaction and thus concentration independent. Recent single-molecule studies from multiple research groups have uncovered that protein concentration can facilitate the unbinding of DNA-bound proteins, revealing regulation of protein unbinding as another mechanistic paradigm for gene regulation. In this Account, we review these recent in vitro and in vivo single-molecule experiments that uncovered the concentration-facilitated protein unbinding by multiple types of DNA-binding proteins, including sequence-nonspecific DNA-binding proteins (e.g., nucleoid-associated proteins, NAP), sequence-specific DNA-binding proteins (e.g., metal-responsive transcription regulators CueR and ZntR), sequence-neutral single-stranded DNA-binding proteins (e.g., Replication protein A, RPA), and DNA polymerases. For the in vitro experiments, Marko's group investigated the exchange of GFP-tagged DNA-bound NAPs with nontagged NAPs in solution of increasing concentration using single-molecule magnetic-tweezers fluorescence microscopy. The faster fluorescence intensity decrease with higher nontagged NAP concentrations suggests that DNA-bound NAPs undergo faster exchange with higher free NAP concentrations. Chen's group used single-molecule fluorescence resonance energy transfer measurements to study the unbinding of CueR from its cognate oligomeric DNA. The average microscopic dwell times of DNA-bound states become shorter with increasing CueR concentrations in the surroundings, demonstrating that free CueR proteins can facilitate the unbinding of the incumbent one on DNA through either assisted dissociation or direct substitution. Greene's group studied the unbinding of RPAs from single-stranded DNA using total internal reflection fluorescence microscopy and DNA curtain techniques. The fluorescence intensity versus time traces show faster decay with higher wild-type RPA concentrations, indicating that DNA-bound RPAs can undergo a concentration-facilitated exchange when encountering excess free RPA. van Oijen's group investigated the leading/lagging-strand polymerase exchange events in the bacteriophage T7 and E. coli replication systems using a combination of single-molecule fluorescence microscopy and DNA-flow-stretching assay. The processivity was observed to have larger decrease when the concentration of the Y526F polymerase mutant increases, indicating that the unbinding of the polymerase is also concentration-dependent. Using stroboscopic imaging and single-molecule tracking, Chen's group further advanced their study into living bacterial cells. They found CueR, as well as its homologue ZntR, shows concentration-enhanced unbinding from its DNA-binding site in vivo. Mechanistic consensus has emerged from these in vitro and in vivo single-molecule studies that encompass a range of proteins with distinct biological functions. It involves multivalent contacts between protein and DNA. The multivalency enables the formation of ternary complexes as intermediates, which subsequently give rise to concentration-enhanced protein unbinding. As multivalent contacts are ubiquitous among DNA-interacting proteins, this multivalency-enabled facilitated unbinding mechanism thus provides a potentially general mechanistic paradigm in regulating protein-DNA interactions.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Direct substitution and assisted dissociation pathways for turning off transcription by a MerR-family metalloregulator.Proc Natl Acad Sci U S A. 2012 Sep 18;109(38):15121-6. doi: 10.1073/pnas.1208508109. Epub 2012 Sep 4. Proc Natl Acad Sci U S A. 2012. PMID: 22949686 Free PMC article.
-
Metalloregulator CueR biases RNA polymerase's kinetic sampling of dead-end or open complex to repress or activate transcription.Proc Natl Acad Sci U S A. 2015 Nov 3;112(44):13467-72. doi: 10.1073/pnas.1515231112. Epub 2015 Oct 19. Proc Natl Acad Sci U S A. 2015. PMID: 26483469 Free PMC article.
-
Facilitated dissociation of nucleoid-associated proteins from DNA in the bacterial confinement.Biophys J. 2022 Apr 5;121(7):1119-1133. doi: 10.1016/j.bpj.2022.03.002. Epub 2022 Mar 5. Biophys J. 2022. PMID: 35257784 Free PMC article.
-
Probing DNA interactions with proteins using a single-molecule toolbox: inside the cell, in a test tube and in a computer.Biochem Soc Trans. 2015 Apr;43(2):139-45. doi: 10.1042/BST20140253. Biochem Soc Trans. 2015. PMID: 26020443 Review.
-
Conformational selection in protein binding and function.Protein Sci. 2014 Nov;23(11):1508-18. doi: 10.1002/pro.2539. Epub 2014 Sep 6. Protein Sci. 2014. PMID: 25155241 Free PMC article. Review.
Cited by
-
Force-Dependent Facilitated Dissociation Can Generate Protein-DNA Catch Bonds.Biophys J. 2019 Sep 17;117(6):1085-1100. doi: 10.1016/j.bpj.2019.07.044. Epub 2019 Aug 2. Biophys J. 2019. PMID: 31427067 Free PMC article.
-
Meddling with Metal Sensors: Fur-Family Proteins as Signaling Hubs.J Bacteriol. 2023 Apr 25;205(4):e0002223. doi: 10.1128/jb.00022-23. Epub 2023 Apr 3. J Bacteriol. 2023. PMID: 37010421 Free PMC article. Review.
-
Driving forces of the complex formation between highly charged disordered proteins.Proc Natl Acad Sci U S A. 2023 Oct 10;120(41):e2304036120. doi: 10.1073/pnas.2304036120. Epub 2023 Oct 5. Proc Natl Acad Sci U S A. 2023. PMID: 37796987 Free PMC article.
-
Receptor-Ligand Rebinding Kinetics in Confinement.Biophys J. 2019 May 7;116(9):1609-1624. doi: 10.1016/j.bpj.2019.02.033. Epub 2019 Apr 5. Biophys J. 2019. PMID: 31029377 Free PMC article.
-
The HMGB chromatin protein Nhp6A can bypass obstacles when traveling on DNA.Nucleic Acids Res. 2020 Nov 4;48(19):10820-10831. doi: 10.1093/nar/gkaa799. Nucleic Acids Res. 2020. PMID: 32997109 Free PMC article.
References
-
- Mueller F, Stasevich TJ, Mazza D, McNally JG. Quantifying transcription factor kinetics: at work or at play? Crit Rev Biochem Mol Biol. 2013;48:492–514. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
