

MERS-CoV spillover at the camel-human interface
- PMID: 29336306
- PMCID: PMC5777824
- DOI: 10.7554/eLife.31257
MERS-CoV spillover at the camel-human interface
Erratum in
-
Correction: MERS-CoV spillover at the camel-human interface.Elife. 2018 Apr 19;7:e37324. doi: 10.7554/eLife.37324. Elife. 2018. PMID: 29669683 Free PMC article. No abstract available.
Abstract
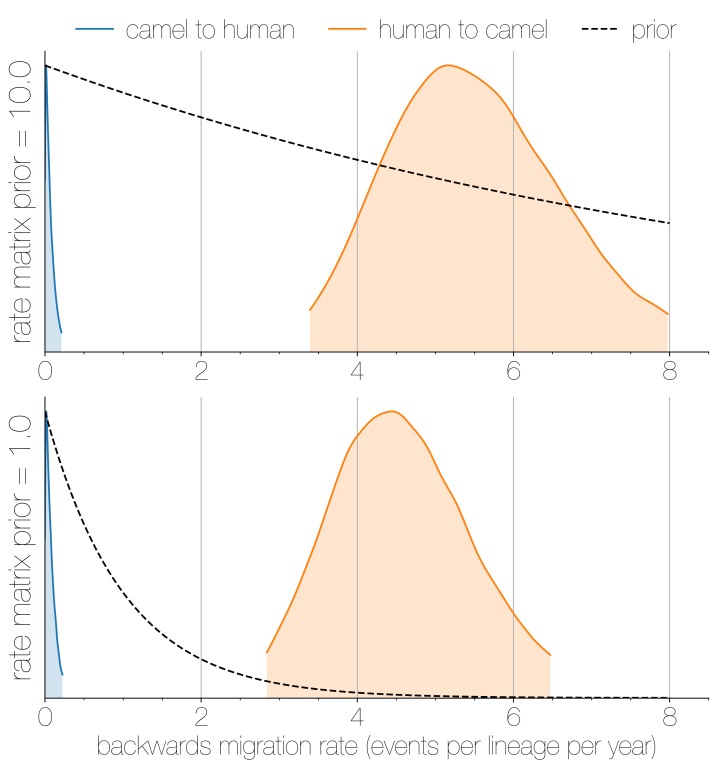
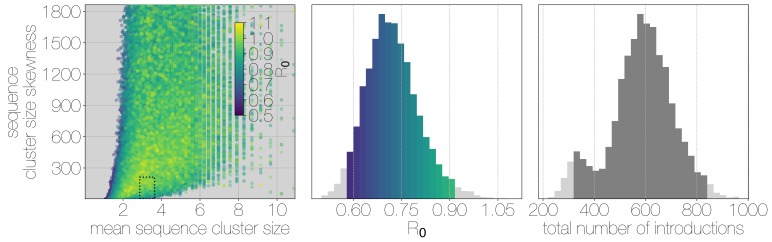
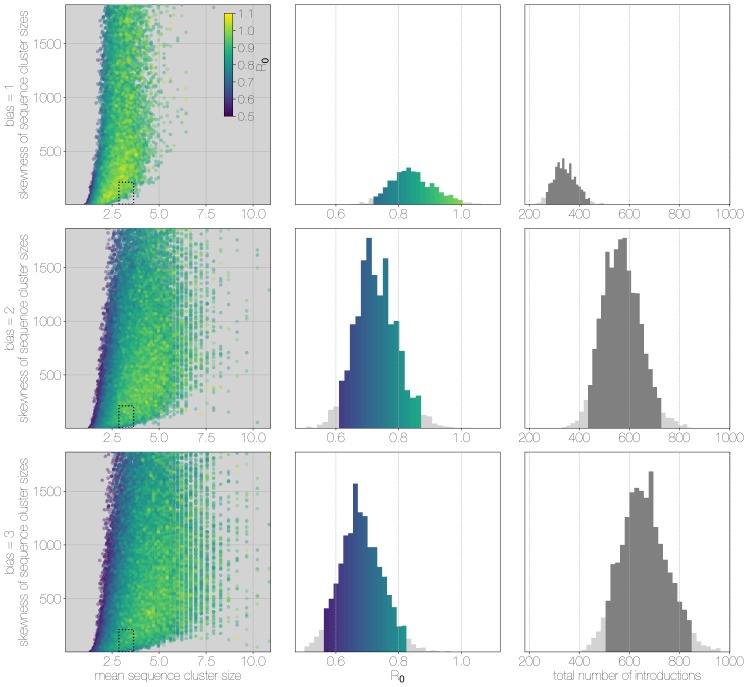
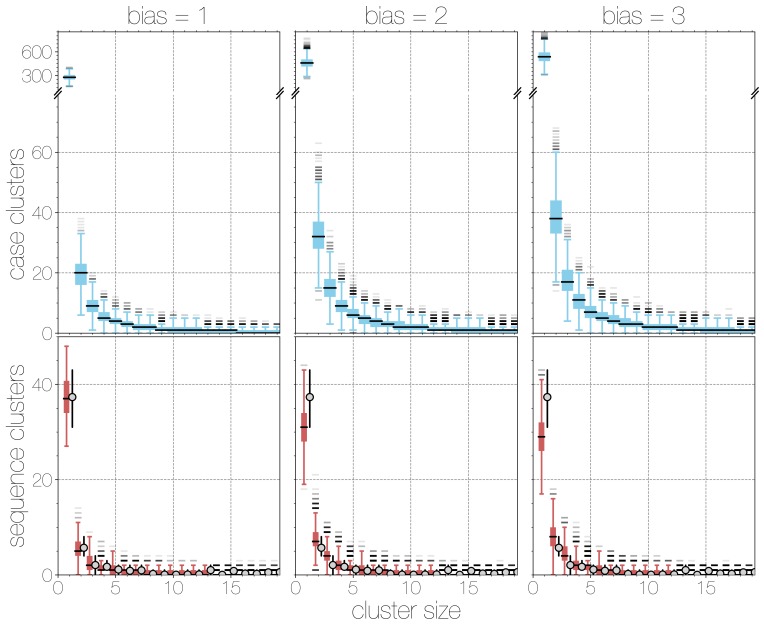
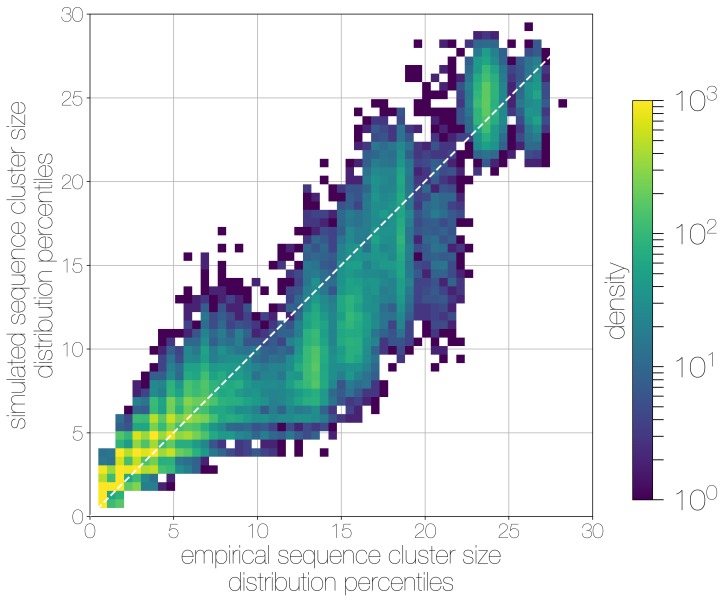
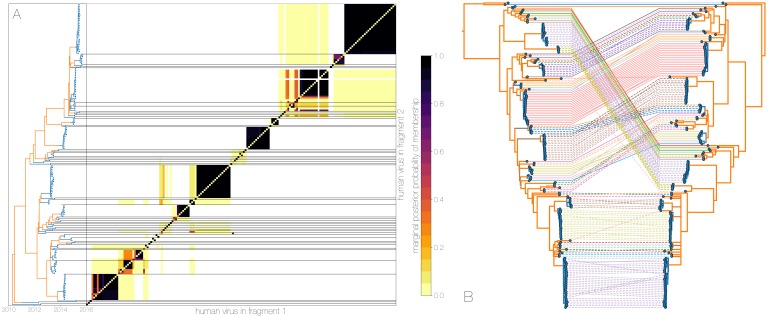
Middle East respiratory syndrome coronavirus (MERS-CoV) is a zoonotic virus from camels causing significant mortality and morbidity in humans in the Arabian Peninsula. The epidemiology of the virus remains poorly understood, and while case-based and seroepidemiological studies have been employed extensively throughout the epidemic, viral sequence data have not been utilised to their full potential. Here, we use existing MERS-CoV sequence data to explore its phylodynamics in two of its known major hosts, humans and camels. We employ structured coalescent models to show that long-term MERS-CoV evolution occurs exclusively in camels, whereas humans act as a transient, and ultimately terminal host. By analysing the distribution of human outbreak cluster sizes and zoonotic introduction times, we show that human outbreaks in the Arabian peninsula are driven by seasonally varying zoonotic transfer of viruses from camels. Without heretofore unseen evolution of host tropism, MERS-CoV is unlikely to become endemic in humans.
Keywords: MERS; coronavirus; epidemiology; evolutionary biology; genomics; global health; phylodynamics; phylogenetics; structured coalescent; virus; zoonosis.
© 2018, Dudas et al.
Conflict of interest statement
GD, LC, AR, TB No competing interests declared
Figures














Similar articles
-
Limited Genetic Diversity Detected in Middle East Respiratory Syndrome-Related Coronavirus Variants Circulating in Dromedary Camels in Jordan.Viruses. 2021 Mar 31;13(4):592. doi: 10.3390/v13040592. Viruses. 2021. PMID: 33807288 Free PMC article.
-
Cross-sectional study of MERS-CoV-specific RNA and antibodies in animals that have had contact with MERS patients in Saudi Arabia.J Infect Public Health. 2018 May-Jun;11(3):331-338. doi: 10.1016/j.jiph.2017.09.022. Epub 2017 Oct 6. J Infect Public Health. 2018. PMID: 28993171 Free PMC article.
-
Genetic diversity and molecular epidemiology of Middle East Respiratory Syndrome Coronavirus in dromedaries in Ethiopia, 2017-2020.Emerg Microbes Infect. 2023 Dec;12(1):e2164218. doi: 10.1080/22221751.2022.2164218. Emerg Microbes Infect. 2023. PMID: 36620913 Free PMC article.
-
MERS coronavirus: diagnostics, epidemiology and transmission.Virol J. 2015 Dec 22;12:222. doi: 10.1186/s12985-015-0439-5. Virol J. 2015. PMID: 26695637 Free PMC article. Review.
-
Evidence for zoonotic origins of Middle East respiratory syndrome coronavirus.J Gen Virol. 2016 Feb;97(2):274-280. doi: 10.1099/jgv.0.000342. Epub 2015 Nov 13. J Gen Virol. 2016. PMID: 26572912 Free PMC article. Review.
Cited by
-
Positive-strand RNA virus replication organelles at a glance.J Cell Sci. 2024 Sep 1;137(17):jcs262164. doi: 10.1242/jcs.262164. Epub 2024 Sep 10. J Cell Sci. 2024. PMID: 39254430 Free PMC article. Review.
-
Recent evolutionary origin and localized diversity hotspots of mammalian coronaviruses.Elife. 2024 Aug 28;13:RP91745. doi: 10.7554/eLife.91745. Elife. 2024. PMID: 39196812 Free PMC article.
-
Middle East Respiratory Syndrome Coronavirus Could be a Priority Pathogen to Cause Public Health Emergency: Noticeable Features and Counteractive Measures.Environ Health Insights. 2024 Aug 15;18:11786302241271545. doi: 10.1177/11786302241271545. eCollection 2024. Environ Health Insights. 2024. PMID: 39156879 Free PMC article.
-
Inactivated Split MERS-CoV Antigen Prevents Lethal Middle East Respiratory Syndrome Coronavirus Infections in Mice.Vaccines (Basel). 2024 Apr 18;12(4):436. doi: 10.3390/vaccines12040436. Vaccines (Basel). 2024. PMID: 38675818 Free PMC article.
-
Genomic Diversity and Recombination Analysis of the Spike Protein Gene from Selected Human Coronaviruses.Biology (Basel). 2024 Apr 22;13(4):282. doi: 10.3390/biology13040282. Biology (Basel). 2024. PMID: 38666894 Free PMC article.
References
-
- Abdallah H, Faye B. Typology of camel farming system in Saudi Arabia. Emirates Journal of Food and Agriculture. 2013;25:250. doi: 10.9755/ejfa.v25i4.15491. - DOI
-
- Ali MA, Shehata MM, Gomaa MR, Kandeil A, El-Shesheny R, Kayed AS, El-Taweel AN, Atea M, Hassan N, Bagato O, Moatasim Y, Mahmoud SH, Kutkat O, Maatouq AM, Osman A, McKenzie PP, Webby RJ, Kayali G, Systematic KG. Systematic, active surveillance for Middle East respiratory syndrome coronavirus in camels in Egypt. Emerging Microbes & Infections. 2017;6:e1. doi: 10.1038/emi.2016.130. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
