

Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus
- PMID: 29190287
- PMCID: PMC5708621
- DOI: 10.1371/journal.ppat.1006698
Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus
Abstract
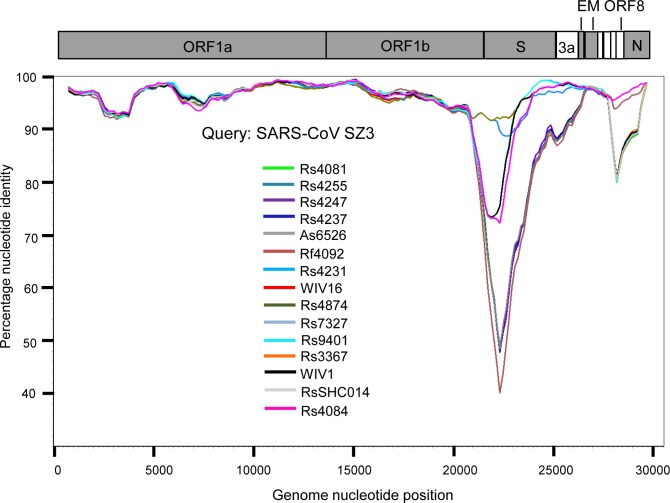
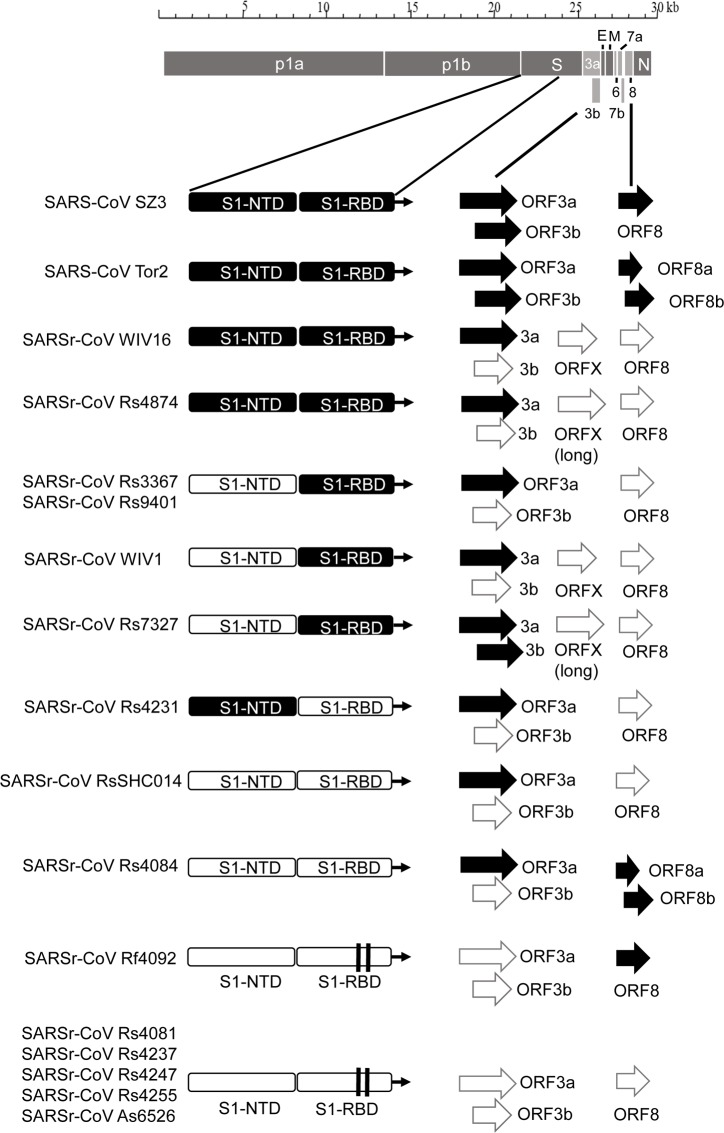
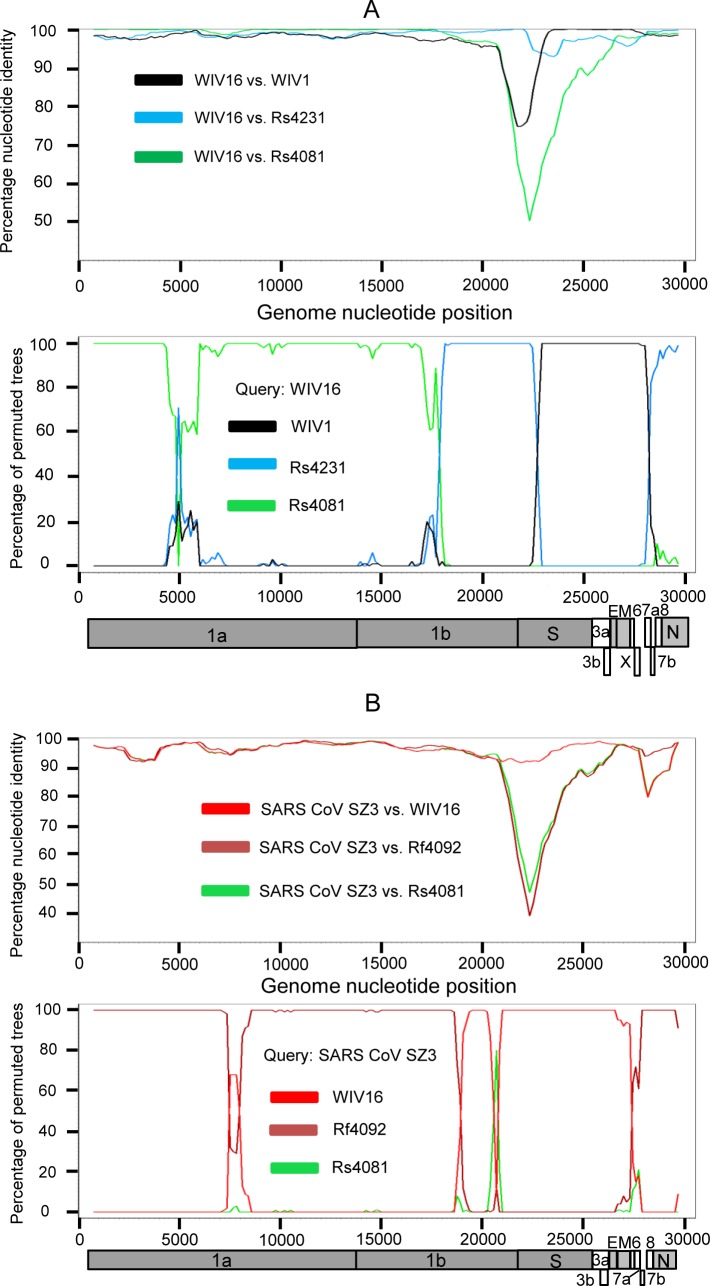
A large number of SARS-related coronaviruses (SARSr-CoV) have been detected in horseshoe bats since 2005 in different areas of China. However, these bat SARSr-CoVs show sequence differences from SARS coronavirus (SARS-CoV) in different genes (S, ORF8, ORF3, etc) and are considered unlikely to represent the direct progenitor of SARS-CoV. Herein, we report the findings of our 5-year surveillance of SARSr-CoVs in a cave inhabited by multiple species of horseshoe bats in Yunnan Province, China. The full-length genomes of 11 newly discovered SARSr-CoV strains, together with our previous findings, reveals that the SARSr-CoVs circulating in this single location are highly diverse in the S gene, ORF3 and ORF8. Importantly, strains with high genetic similarity to SARS-CoV in the hypervariable N-terminal domain (NTD) and receptor-binding domain (RBD) of the S1 gene, the ORF3 and ORF8 region, respectively, were all discovered in this cave. In addition, we report the first discovery of bat SARSr-CoVs highly similar to human SARS-CoV in ORF3b and in the split ORF8a and 8b. Moreover, SARSr-CoV strains from this cave were more closely related to SARS-CoV in the non-structural protein genes ORF1a and 1b compared with those detected elsewhere. Recombination analysis shows evidence of frequent recombination events within the S gene and around the ORF8 between these SARSr-CoVs. We hypothesize that the direct progenitor of SARS-CoV may have originated after sequential recombination events between the precursors of these SARSr-CoVs. Cell entry studies demonstrated that three newly identified SARSr-CoVs with different S protein sequences are all able to use human ACE2 as the receptor, further exhibiting the close relationship between strains in this cave and SARS-CoV. This work provides new insights into the origin and evolution of SARS-CoV and highlights the necessity of preparedness for future emergence of SARS-like diseases.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures






Comment in
-
Don't misrepresent link between bats and SARS.Nature. 2018 Jan 18;553(7688):281. doi: 10.1038/d41586-018-00603-7. Nature. 2018. PMID: 29345663 No abstract available.
Similar articles
-
Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination.J Virol. 2015 Oct;89(20):10532-47. doi: 10.1128/JVI.01048-15. Epub 2015 Aug 12. J Virol. 2015. PMID: 26269185 Free PMC article.
-
Epidemiology and Genomic Characterization of Two Novel SARS-Related Coronaviruses in Horseshoe Bats from Guangdong, China.mBio. 2022 Jun 28;13(3):e0046322. doi: 10.1128/mbio.00463-22. Epub 2022 Apr 25. mBio. 2022. PMID: 35467426 Free PMC article.
-
Molecular epidemiology, evolution and phylogeny of SARS coronavirus.Infect Genet Evol. 2019 Jul;71:21-30. doi: 10.1016/j.meegid.2019.03.001. Epub 2019 Mar 4. Infect Genet Evol. 2019. PMID: 30844511 Free PMC article. Review.
-
Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events.J Virol. 2010 Mar;84(6):2808-19. doi: 10.1128/JVI.02219-09. Epub 2010 Jan 13. J Virol. 2010. PMID: 20071579 Free PMC article.
-
Geographical structure of bat SARS-related coronaviruses.Infect Genet Evol. 2019 Apr;69:224-229. doi: 10.1016/j.meegid.2019.02.001. Epub 2019 Feb 6. Infect Genet Evol. 2019. PMID: 30735813 Free PMC article. Review.
Cited by
-
Identification and Characterization of an Alphacoronavirus in Rhinolophus sinicus and a Betacoronavirus in Apodemus ilex in Yunnan, China.Microorganisms. 2024 Jul 21;12(7):1490. doi: 10.3390/microorganisms12071490. Microorganisms. 2024. PMID: 39065258 Free PMC article.
-
Comparative Review of SARS-CoV-2, SARS-CoV, MERS-CoV, and Influenza A Respiratory Viruses.Front Immunol. 2020 Sep 11;11:552909. doi: 10.3389/fimmu.2020.552909. eCollection 2020. Front Immunol. 2020. PMID: 33013925 Free PMC article. Review.
-
Phylogeography, Transmission, and Viral Proteins of Nipah Virus.Virol Sin. 2018 Oct;33(5):385-393. doi: 10.1007/s12250-018-0050-1. Epub 2018 Oct 11. Virol Sin. 2018. PMID: 30311101 Free PMC article. Review.
-
Viral Hyperparasitism in Bat Ectoparasites: Implications for Pathogen Maintenance and Transmission.Microorganisms. 2022 Jun 16;10(6):1230. doi: 10.3390/microorganisms10061230. Microorganisms. 2022. PMID: 35744747 Free PMC article. Review.
-
The papain-like protease determines a virulence trait that varies among members of the SARS-coronavirus species.PLoS Pathog. 2018 Sep 24;14(9):e1007296. doi: 10.1371/journal.ppat.1007296. eCollection 2018 Sep. PLoS Pathog. 2018. PMID: 30248143 Free PMC article.
References
-
- Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med. 2004; 10: S88–97. doi: 10.1038/nm1143 - DOI - PMC - PubMed
-
- Chinese SMEC. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004; 303: 1666–1669. doi: 10.1126/science.1092002 - DOI - PubMed
-
- Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 2014; 101: 45–56. doi: 10.1016/j.antiviral.2013.10.013 - DOI - PMC - PubMed
-
- Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, et al. The Genome sequence of the SARS-associated coronavirus. Science. 2003; 300: 1399–1404. doi: 10.1126/science.1085953 - DOI - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
