

The skeletal phenotype of achondrogenesis type 1A is caused exclusively by cartilage defects
- PMID: 29180569
- PMCID: PMC5825869
- DOI: 10.1242/dev.156588
The skeletal phenotype of achondrogenesis type 1A is caused exclusively by cartilage defects
Abstract
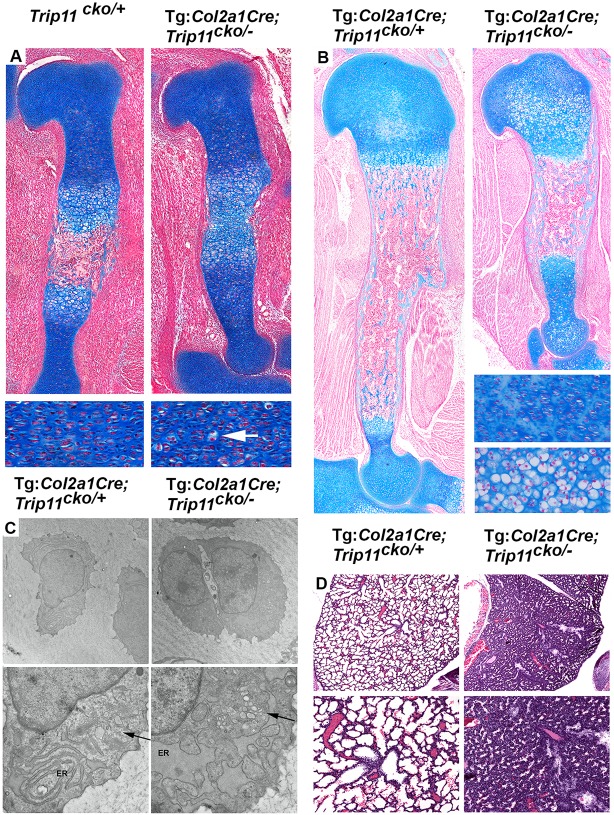
Inactivating mutations in the ubiquitously expressed membrane trafficking component GMAP-210 (encoded by Trip11) cause achondrogenesis type 1A (ACG1A). ACG1A is surprisingly tissue specific, mainly affecting cartilage development. Bone development is also abnormal, but as chondrogenesis and osteogenesis are closely coupled, this could be a secondary consequence of the cartilage defect. A possible explanation for the tissue specificity of ACG1A is that cartilage and bone are highly secretory tissues with a high use of the membrane trafficking machinery. The perinatal lethality of ACG1A prevents investigating this hypothesis. We therefore generated mice with conditional Trip11 knockout alleles and inactivated Trip11 in chondrocytes, osteoblasts, osteoclasts and pancreas acinar cells, all highly secretory cell types. We discovered that the ACG1A skeletal phenotype is solely due to absence of GMAP-210 in chondrocytes. Mice lacking GMAP-210 in osteoblasts, osteoclasts and acinar cells were normal. When we inactivated Trip11 in primary chondrocyte cultures, GMAP-210 deficiency affected trafficking of a subset of chondrocyte-expressed proteins rather than globally impairing membrane trafficking. Thus, GMAP-210 is essential for trafficking specific cargoes in chondrocytes but is dispensable in other highly secretory cells.
Keywords: Achondrogenesis type 1A; Cartilage; Conditional knockout; GMAP-210; Golgin; Proteomics.
© 2018. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interestsThe authors declare no competing or financial interests.
Figures








Similar articles
-
A common pathomechanism in GMAP-210- and LBR-related diseases.JCI Insight. 2018 Dec 6;3(23):e121150. doi: 10.1172/jci.insight.121150. JCI Insight. 2018. PMID: 30518689 Free PMC article.
-
Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210.N Engl J Med. 2010 Jan 21;362(3):206-16. doi: 10.1056/NEJMoa0900158. N Engl J Med. 2010. PMID: 20089971 Free PMC article.
-
Biallelic deep intronic variant c.5457+81T>A in TRIP11 causes loss of function and results in achondrogenesis 1A.Hum Mutat. 2021 Aug;42(8):1005-1014. doi: 10.1002/humu.24235. Epub 2021 Jun 8. Hum Mutat. 2021. PMID: 34057271
-
Genetic control of skeletal development.Curr Opin Genet Dev. 2001 Oct;11(5):527-32. doi: 10.1016/s0959-437x(00)00228-8. Curr Opin Genet Dev. 2001. PMID: 11532394 Review.
-
Signaling pathways effecting crosstalk between cartilage and adjacent tissues: Seminars in cell and developmental biology: The biology and pathology of cartilage.Semin Cell Dev Biol. 2017 Feb;62:16-33. doi: 10.1016/j.semcdb.2016.05.007. Epub 2016 May 12. Semin Cell Dev Biol. 2017. PMID: 27180955 Review.
Cited by
-
Disruption of Trip11 in cranial neural crest cells is associated with increased ER and Golgi stress contributing to skull defects in mice.Dev Dyn. 2022 Jul;251(7):1209-1222. doi: 10.1002/dvdy.461. Epub 2022 Feb 18. Dev Dyn. 2022. PMID: 35147267 Free PMC article.
-
A common pathomechanism in GMAP-210- and LBR-related diseases.JCI Insight. 2018 Dec 6;3(23):e121150. doi: 10.1172/jci.insight.121150. JCI Insight. 2018. PMID: 30518689 Free PMC article.
-
Different Forms of ER Stress in Chondrocytes Result in Short Stature Disorders and Degenerative Cartilage Diseases: New Insights by Cartilage-Specific ERp57 Knockout Mice.Oxid Med Cell Longev. 2018 Dec 17;2018:8421394. doi: 10.1155/2018/8421394. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 30647818 Free PMC article. Review.
-
The factory, the antenna and the scaffold: the three-way interplay between the Golgi, cilium and extracellular matrix underlying tissue function.Biol Open. 2023 Feb 15;12(2):bio059719. doi: 10.1242/bio.059719. Epub 2023 Feb 21. Biol Open. 2023. PMID: 36802341 Free PMC article. Review.
-
The Physiological Functions of the Golgin Vesicle Tethering Proteins.Front Cell Dev Biol. 2019 Jun 18;7:94. doi: 10.3389/fcell.2019.00094. eCollection 2019. Front Cell Dev Biol. 2019. PMID: 31316978 Free PMC article. Review.
References
-
- Benjamini Y. and Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B 57, 289-300.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
