

Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions
- PMID: 29170434
- PMCID: PMC5700980
- DOI: 10.1038/s41467-017-01962-x
Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions
Abstract
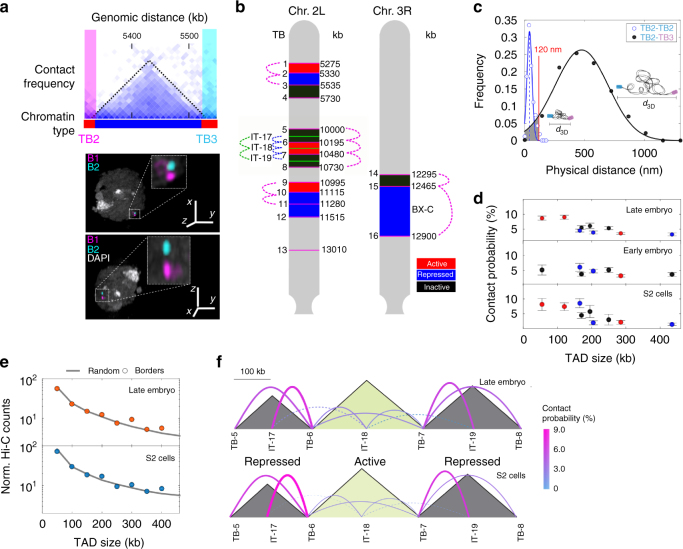
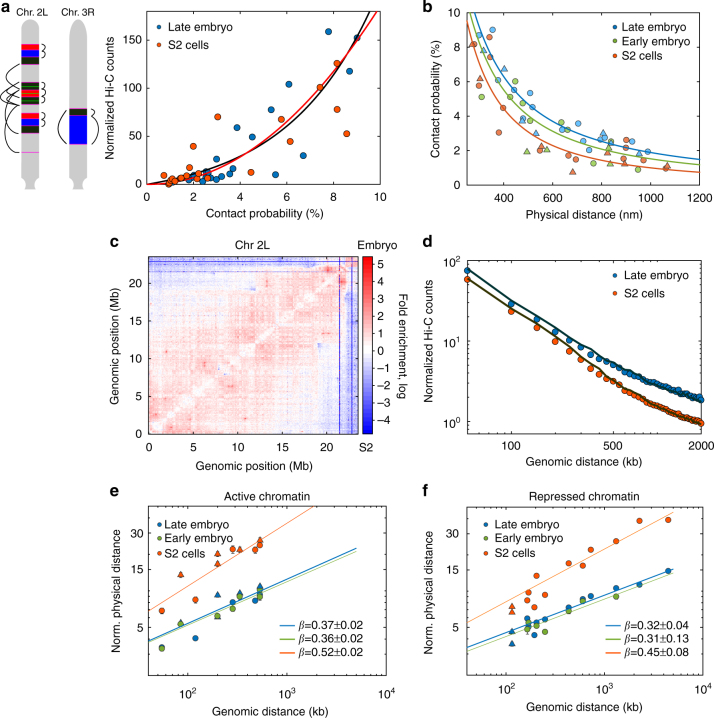
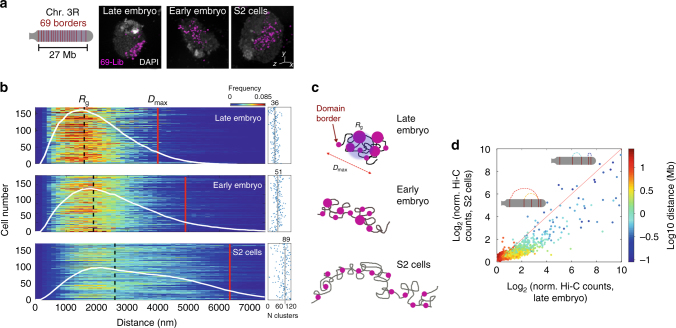
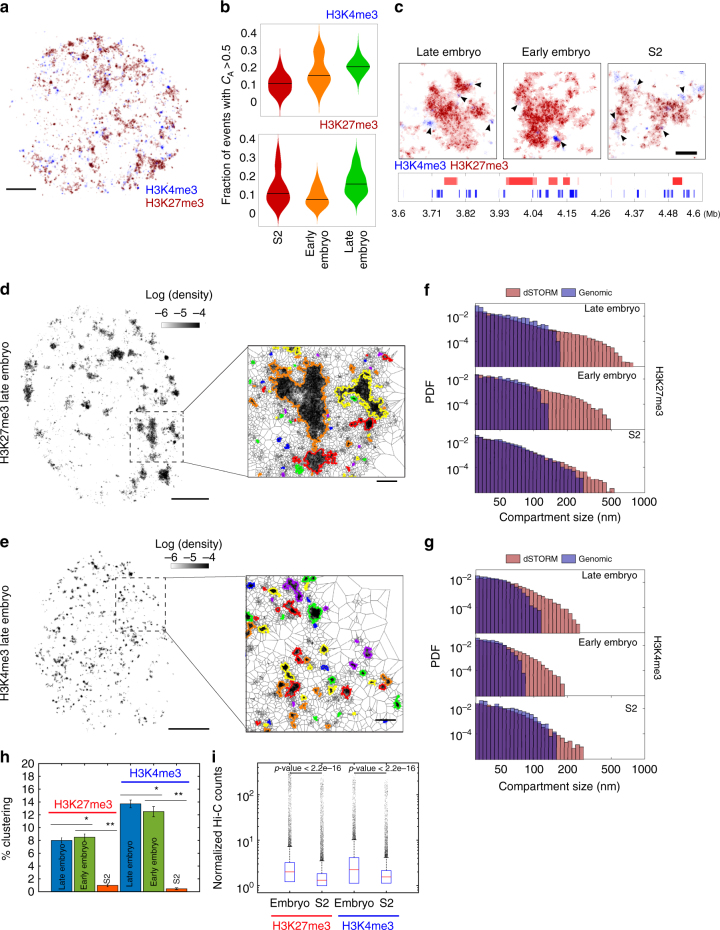
At the kilo- to megabase pair scales, eukaryotic genomes are partitioned into self-interacting modules or topologically associated domains (TADs) that associate to form nuclear compartments. Here, we combine high-content super-resolution microscopies with state-of-the-art DNA-labeling methods to reveal the variability in the multiscale organization of the Drosophila genome. We find that association frequencies within TADs and between TAD borders are below ~10%, independently of TAD size, epigenetic state, or cell type. Critically, despite this large heterogeneity, we are able to visualize nanometer-sized epigenetic domains at the single-cell level. In addition, absolute contact frequencies within and between TADs are to a large extent defined by genomic distance, higher-order chromosome architecture, and epigenetic identity. We propose that TADs and compartments are organized by multiple, small-frequency, yet specific interactions that are regulated by epigenetics and transcriptional state.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation.Mol Syst Biol. 2015 Dec 23;11(12):852. doi: 10.15252/msb.20156492. Mol Syst Biol. 2015. PMID: 26700852 Free PMC article.
-
MrTADFinder: A network modularity based approach to identify topologically associating domains in multiple resolutions.PLoS Comput Biol. 2017 Jul 24;13(7):e1005647. doi: 10.1371/journal.pcbi.1005647. eCollection 2017 Jul. PLoS Comput Biol. 2017. PMID: 28742097 Free PMC article.
-
TADs are 3D structural units of higher-order chromosome organization in Drosophila.Sci Adv. 2018 Feb 28;4(2):eaar8082. doi: 10.1126/sciadv.aar8082. eCollection 2018 Feb. Sci Adv. 2018. PMID: 29503869 Free PMC article.
-
Principles of genome folding into topologically associating domains.Sci Adv. 2019 Apr 10;5(4):eaaw1668. doi: 10.1126/sciadv.aaw1668. eCollection 2019 Apr. Sci Adv. 2019. PMID: 30989119 Free PMC article. Review.
-
Are TADs supercoiled?Nucleic Acids Res. 2019 Jan 25;47(2):521-532. doi: 10.1093/nar/gky1091. Nucleic Acids Res. 2019. PMID: 30395328 Free PMC article. Review.
Cited by
-
Independence of chromatin conformation and gene regulation during Drosophila dorsoventral patterning.Nat Genet. 2021 Apr;53(4):487-499. doi: 10.1038/s41588-021-00799-x. Epub 2021 Apr 1. Nat Genet. 2021. PMID: 33795866 Free PMC article.
-
A Shift in Paradigms: Spatial Genomics Approaches to Reveal Single-Cell Principles of Genome Organization.Front Genet. 2021 Nov 19;12:780822. doi: 10.3389/fgene.2021.780822. eCollection 2021. Front Genet. 2021. PMID: 34868269 Free PMC article. Review.
-
Advances in Chromatin Imaging at Kilobase-Scale Resolution.Trends Genet. 2020 Apr;36(4):273-287. doi: 10.1016/j.tig.2019.12.010. Epub 2020 Jan 29. Trends Genet. 2020. PMID: 32007290 Free PMC article. Review.
-
Polymer modelling unveils the roles of heterochromatin and nucleolar organizing regions in shaping 3D genome organization in Arabidopsis thaliana.Nucleic Acids Res. 2021 Feb 26;49(4):1840-1858. doi: 10.1093/nar/gkaa1275. Nucleic Acids Res. 2021. PMID: 33444439 Free PMC article.
-
Revisiting the organization of Polycomb-repressed domains: 3D chromatin models from Hi-C compared with super-resolution imaging.Nucleic Acids Res. 2020 Nov 18;48(20):11486-11494. doi: 10.1093/nar/gkaa932. Nucleic Acids Res. 2020. PMID: 33095877 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
