

Homology-based hydrogen bond information improves crystallographic structures in the PDB
- PMID: 29168245
- PMCID: PMC5818736
- DOI: 10.1002/pro.3353
Homology-based hydrogen bond information improves crystallographic structures in the PDB
Abstract
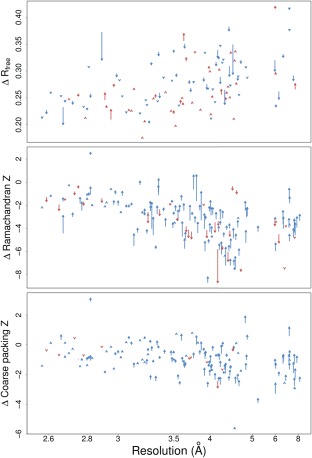
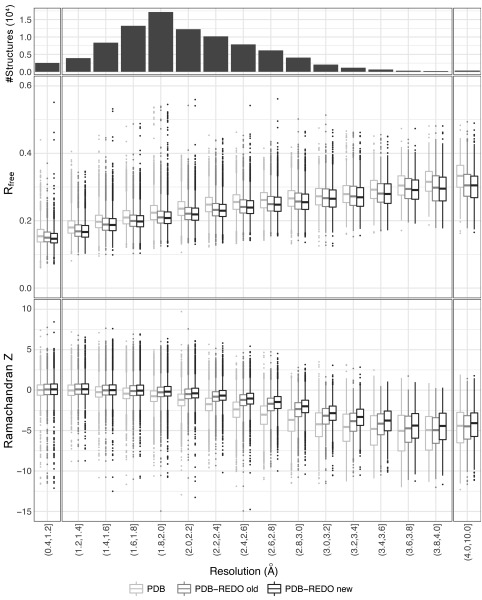
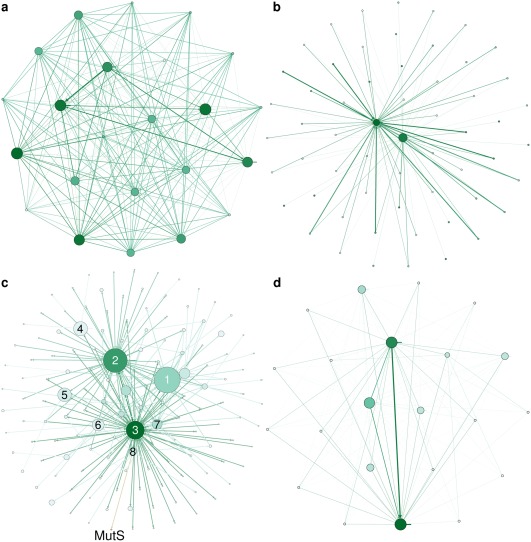
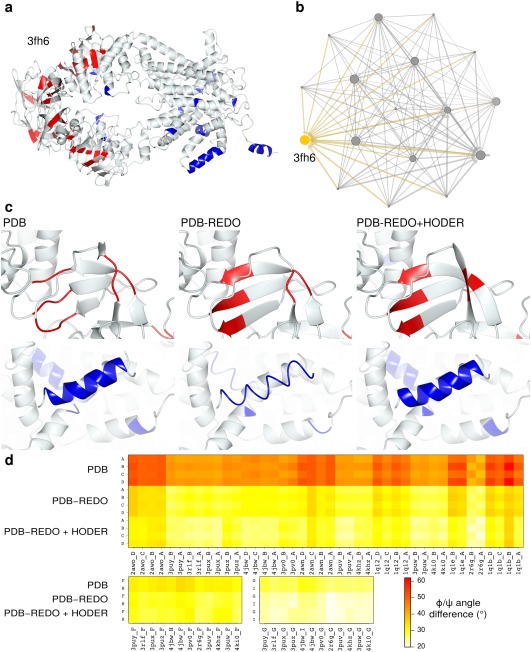
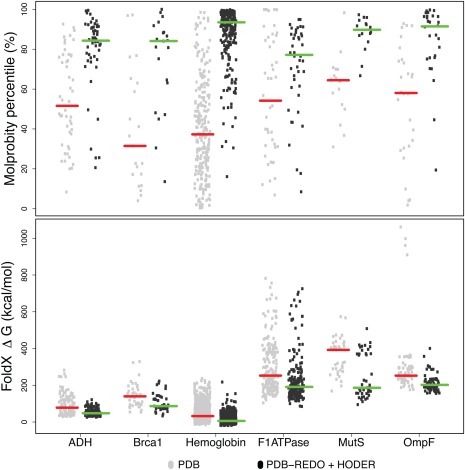
The Protein Data Bank (PDB) is the global archive for structural information on macromolecules, and a popular resource for researchers, teachers, and students, amassing more than one million unique users each year. Crystallographic structure models in the PDB (more than 100,000 entries) are optimized against the crystal diffraction data and geometrical restraints. This process of crystallographic refinement typically ignored hydrogen bond (H-bond) distances as a source of information. However, H-bond restraints can improve structures at low resolution where diffraction data are limited. To improve low-resolution structure refinement, we present methods for deriving H-bond information either globally from well-refined high-resolution structures from the PDB-REDO databank, or specifically from on-the-fly constructed sets of homologous high-resolution structures. Refinement incorporating HOmology DErived Restraints (HODER), improves geometrical quality and the fit to the diffraction data for many low-resolution structures. To make these improvements readily available to the general public, we applied our new algorithms to all crystallographic structures in the PDB: using massively parallel computing, we constructed a new instance of the PDB-REDO databank (https://pdb-redo.eu). This resource is useful for researchers to gain insight on individual structures, on specific protein families (as we demonstrate with examples), and on general features of protein structure using data mining approaches on a uniformly treated dataset.
Keywords: PDB; PDB-REDO; X-ray crystallography; databank; high-throughput computing; homology; hydrogen bonds; refinement; restraints.
© 2017 The Protein Society.
Figures
Similar articles
-
Automatic rebuilding and optimization of crystallographic structures in the Protein Data Bank.Bioinformatics. 2011 Dec 15;27(24):3392-8. doi: 10.1093/bioinformatics/btr590. Epub 2011 Oct 27. Bioinformatics. 2011. PMID: 22034521 Free PMC article.
-
Neutron crystallographic refinement with REFMAC5 from the CCP4 suite.Acta Crystallogr D Struct Biol. 2023 Dec 1;79(Pt 12):1056-1070. doi: 10.1107/S2059798323008793. Epub 2023 Nov 3. Acta Crystallogr D Struct Biol. 2023. PMID: 37921806 Free PMC article.
-
Homology-based loop modeling yields more complete crystallographic protein structures.IUCrJ. 2018 Aug 8;5(Pt 5):585-594. doi: 10.1107/S2052252518010552. eCollection 2018 Sep 1. IUCrJ. 2018. PMID: 30224962 Free PMC article.
-
New Biological Insights from Better Structure Models.J Mol Biol. 2016 Mar 27;428(6):1375-1393. doi: 10.1016/j.jmb.2016.02.002. Epub 2016 Feb 8. J Mol Biol. 2016. PMID: 26869101 Review.
-
Protein Data Bank: A Comprehensive Review of 3D Structure Holdings and Worldwide Utilization by Researchers, Educators, and Students.Biomolecules. 2022 Oct 4;12(10):1425. doi: 10.3390/biom12101425. Biomolecules. 2022. PMID: 36291635 Free PMC article. Review.
Cited by
-
Updated restraint dictionaries for carbohydrates in the pyranose form.Acta Crystallogr D Struct Biol. 2022 Apr 1;78(Pt 4):455-465. doi: 10.1107/S2059798322001103. Epub 2022 Mar 4. Acta Crystallogr D Struct Biol. 2022. PMID: 35362468 Free PMC article.
-
The use of local structural similarity of distant homologues for crystallographic model building from a molecular-replacement solution.Acta Crystallogr D Struct Biol. 2020 Mar 1;76(Pt 3):248-260. doi: 10.1107/S2059798320000455. Epub 2020 Feb 28. Acta Crystallogr D Struct Biol. 2020. PMID: 32133989 Free PMC article.
-
Towards Consistency in Geometry Restraints for Carbohydrates in the Pyranose form: Modern Dictionary Generators Reviewed.Curr Med Chem. 2022;29(7):1193-1207. doi: 10.2174/0929867328666210902140754. Curr Med Chem. 2022. PMID: 34477506 Free PMC article.
-
Making glycoproteins a little bit sweeter with PDB-REDO.Acta Crystallogr F Struct Biol Commun. 2018 Aug 1;74(Pt 8):463-472. doi: 10.1107/S2053230X18004016. Epub 2018 Jul 26. Acta Crystallogr F Struct Biol Commun. 2018. PMID: 30084395 Free PMC article.
-
The structure of nontypeable Haemophilus influenzae SapA in a closed conformation reveals a constricted ligand-binding cavity and a novel RNA binding motif.PLoS One. 2021 Oct 15;16(10):e0256070. doi: 10.1371/journal.pone.0256070. eCollection 2021. PLoS One. 2021. PMID: 34653190 Free PMC article.
References
-
- Kleywegt GJ, Jones TA (2002) Homo crystallographicus ‐ quo vadis? Structure 10:465–472. - PubMed
-
- Engh RA, Huber R (1991) Accurate bond and angle parameters for X‐ray protein structure refinement. Acta Cryst 47:392–400.
-
- Parkinson G, Vojtechovsky J, Clowney L, Brünger AT, Berman HM (1996) New parameters for the refinement of nucleic acid‐containing structures. Acta Cryst 52:57–64. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
