

Molecular mechanisms of programmed cell death-1 dependent T cell suppression: relevance for immunotherapy
- PMID: 29114543
- PMCID: PMC5653513
- DOI: 10.21037/atm.2017.06.11
Molecular mechanisms of programmed cell death-1 dependent T cell suppression: relevance for immunotherapy
Abstract
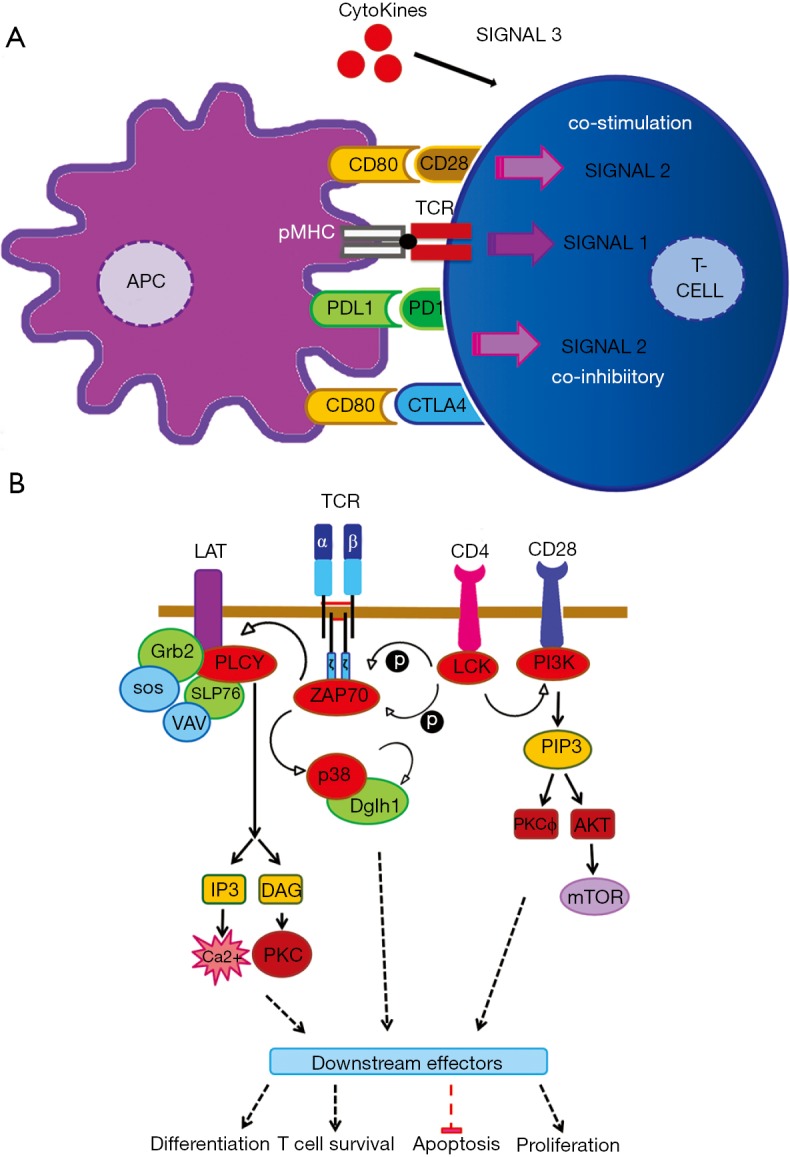
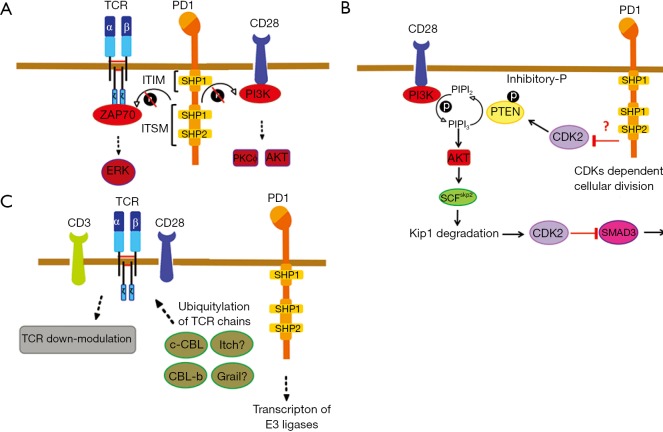
Programmed cell death-1 (PD1) has become a significant target for cancer immunotherapy. PD1 and its receptor programmed cell death 1 ligand 1 (PDL1) are key regulatory physiological immune checkpoints that maintain self-tolerance in the organism by regulating the degree of activation of T and B cells amongst other immune cell types. However, cancer cells take advantage of these immunosuppressive regulatory mechanisms to escape T and B cell-mediated immunity. PD1 engagement on T cells by PDL1 on the surface of cancer cells dramatically interferes with T cell activation and the acquisition of effector capacities. Interestingly, PD1-targeted therapies have demonstrated to be highly effective in rescuing T cell anti-tumor effector functions. Amongst these the use of anti-PD1/PDL1 monoclonal antibodies are particularly efficacious in human therapies. Furthermore, clinical findings with PD1/PDL1 blockers over several cancer types demonstrate clinical benefit. Despite the successful results, the molecular mechanisms by which PD1-targeted therapies rescue T cell functions still remain elusive. Therefore, it is a key issue to uncover the molecular pathways by which these therapies exert its function in T cells. A profound knowledge of PDL1/PD1 mechanisms will surely uncover a new array of targets susceptible of therapeutic intervention. Here, we provide an overview of the molecular events underlying PD1-dependent T cell suppression in cancer.
Keywords: Cancer; immune checkpoint inhibitors; immunotherapy.
Conflict of interest statement
Conflicts of Interest: The authors have no conflicts of interest to declare.
Figures

Similar articles
-
[Immunotherapy in non-small cell lung cancer: inhibition of PD1/PDL1 pathway].Rev Pneumol Clin. 2015 Feb;71(1):44-56. doi: 10.1016/j.pneumo.2014.11.004. Epub 2015 Feb 14. Rev Pneumol Clin. 2015. PMID: 25687821 Review. French.
-
PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy.Front Pharmacol. 2021 Sep 1;12:731798. doi: 10.3389/fphar.2021.731798. eCollection 2021. Front Pharmacol. 2021. PMID: 34539412 Free PMC article. Review.
-
Macrocyclic Compounds from Ansamycin Antibiotic Class as Inhibitors of PD1-PDL1 Protein-Protein Interaction.Chem Pharm Bull (Tokyo). 2018;66(8):773-778. doi: 10.1248/cpb.c17-00800. Chem Pharm Bull (Tokyo). 2018. PMID: 30068796
-
Trends in the Research Into Immune Checkpoint Blockade by Anti-PD1/PDL1 Antibodies in Cancer Immunotherapy: A Bibliometric Study.Front Pharmacol. 2021 Aug 17;12:670900. doi: 10.3389/fphar.2021.670900. eCollection 2021. Front Pharmacol. 2021. PMID: 34489691 Free PMC article.
-
The advantages of PD1 activating chimeric receptor (PD1-ACR) engineered lymphocytes for PDL1(+) cancer therapy.Am J Transl Res. 2015 Mar 15;7(3):460-73. eCollection 2015. Am J Transl Res. 2015. PMID: 26045887 Free PMC article.
Cited by
-
Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy.Cells. 2020 Dec 24;10(1):14. doi: 10.3390/cells10010014. Cells. 2020. PMID: 33374128 Free PMC article. Review.
-
Prognostic Value of BRAF, Programmed Cell Death 1 (PD1), and PD Ligand 1 (PDL1) Protein Expression in Colon Adenocarcinoma.Diagnostics (Basel). 2023 Jan 9;13(2):237. doi: 10.3390/diagnostics13020237. Diagnostics (Basel). 2023. PMID: 36673047 Free PMC article.
-
PDL1 shapes the classical Hodgkin lymphoma microenvironment without inducing T-cell exhaustion.Haematologica. 2023 Apr 1;108(4):1068-1082. doi: 10.3324/haematol.2022.280014. Haematologica. 2023. PMID: 35833296 Free PMC article.
-
The biology and therapeutic management of melanoma brain metastases.Biochem Pharmacol. 2018 Jul;153:35-45. doi: 10.1016/j.bcp.2017.12.019. Epub 2017 Dec 24. Biochem Pharmacol. 2018. PMID: 29278675 Free PMC article. Review.
-
Immune Checkpoint Molecules in Primary Diffuse Large B-Cell Lymphoma of the Central Nervous System.Basic Clin Neurosci. 2020 Jul-Aug;11(4):491-498. doi: 10.32598/bcn.11.4.2542.1. Epub 2020 Jul 1. Basic Clin Neurosci. 2020. PMID: 33613887 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials