

Modulators of Redox Metabolism in Head and Neck Cancer
- PMID: 29113454
- PMCID: PMC6207163
- DOI: 10.1089/ars.2017.7423
Modulators of Redox Metabolism in Head and Neck Cancer
Abstract
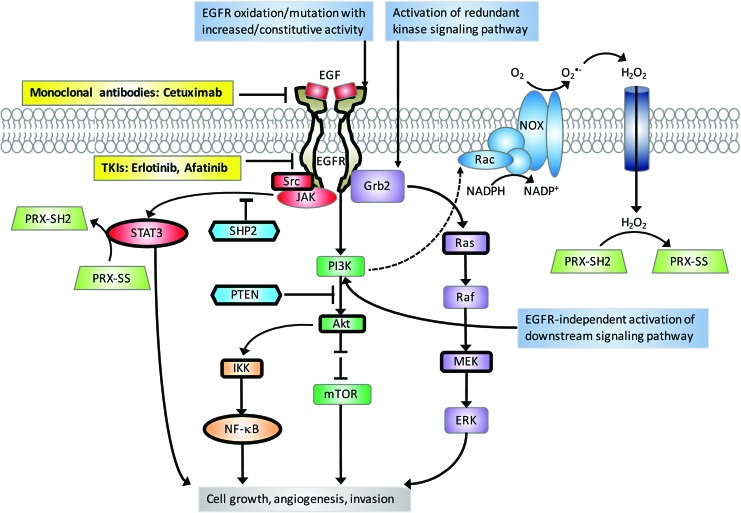
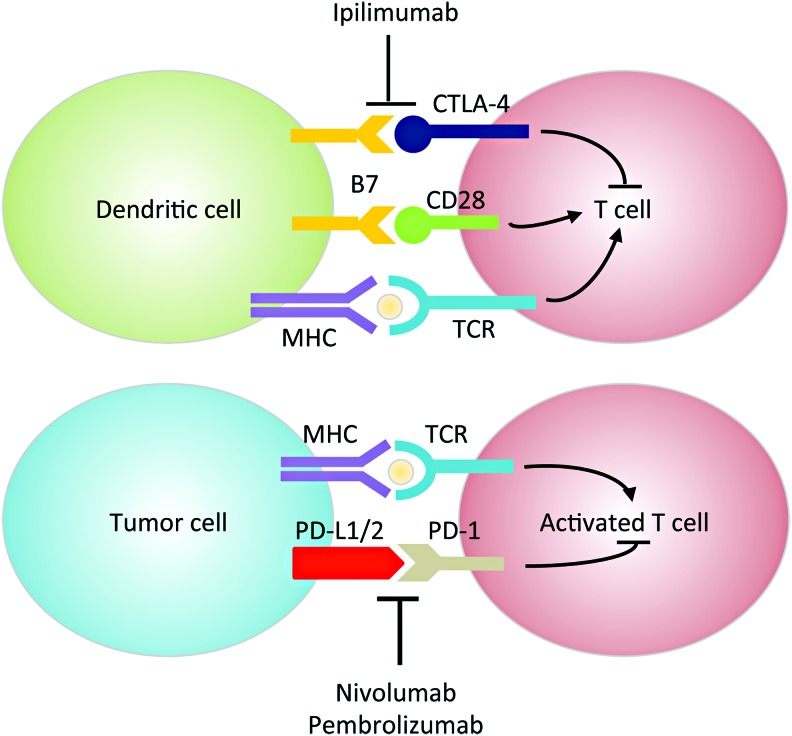
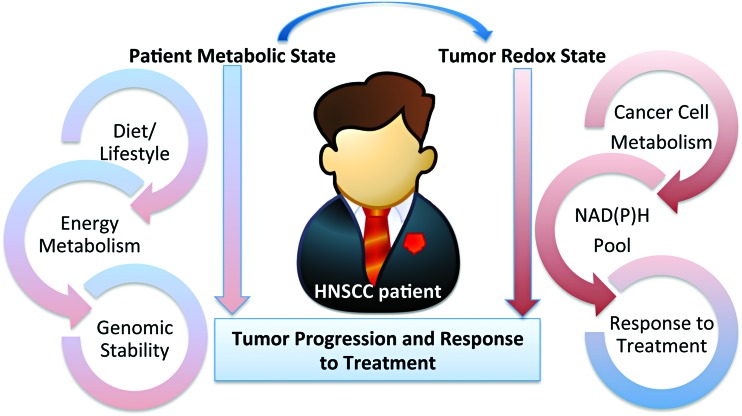
Significance: Head and neck squamous cell cancer (HNSCC) is a complex disease characterized by high genetic and metabolic heterogeneity. Radiation therapy (RT) alone or combined with systemic chemotherapy is widely used for treatment of HNSCC as definitive treatment or as adjuvant treatment after surgery. Antibodies against epidermal growth factor receptor are used in definitive or palliative treatment. Recent Advances: Emerging targeted therapies against other proteins of interest as well as programmed cell death protein 1 and programmed death-ligand 1 immunotherapies are being explored in clinical trials.
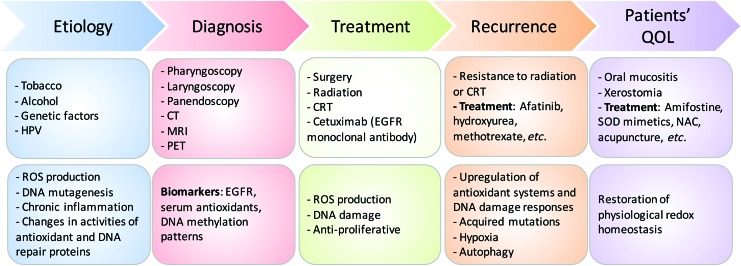
Critical issues: The disease heterogeneity, invasiveness, and resistance to standard of care RT or chemoradiation therapy continue to constitute significant roadblocks for treatment and patients' quality of life (QOL) despite improvements in treatment modality and the emergence of new therapies over the past two decades.
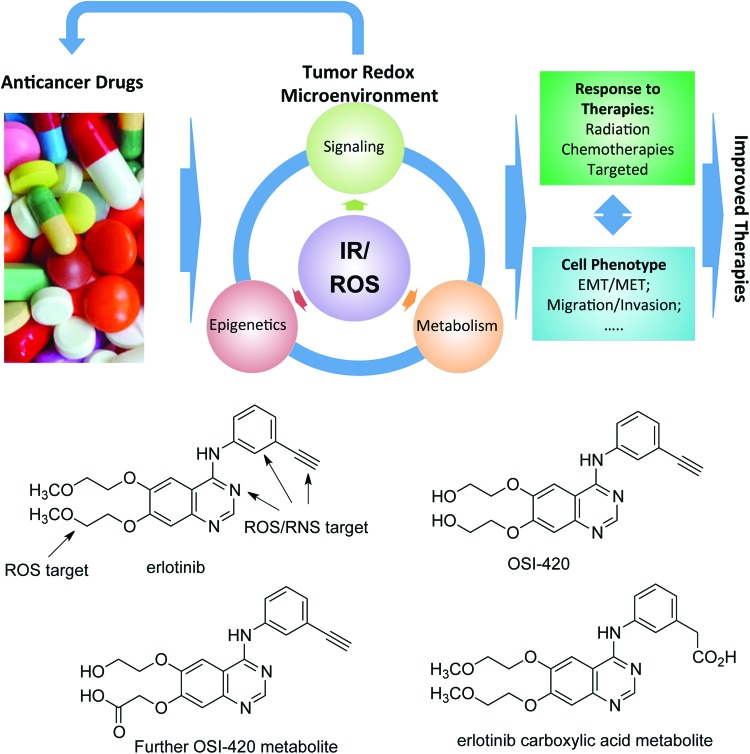
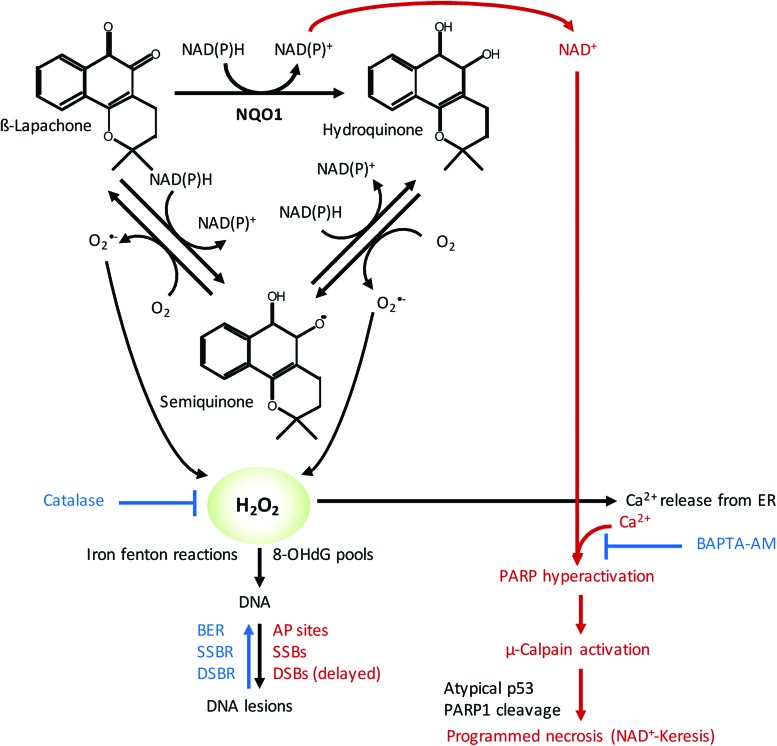
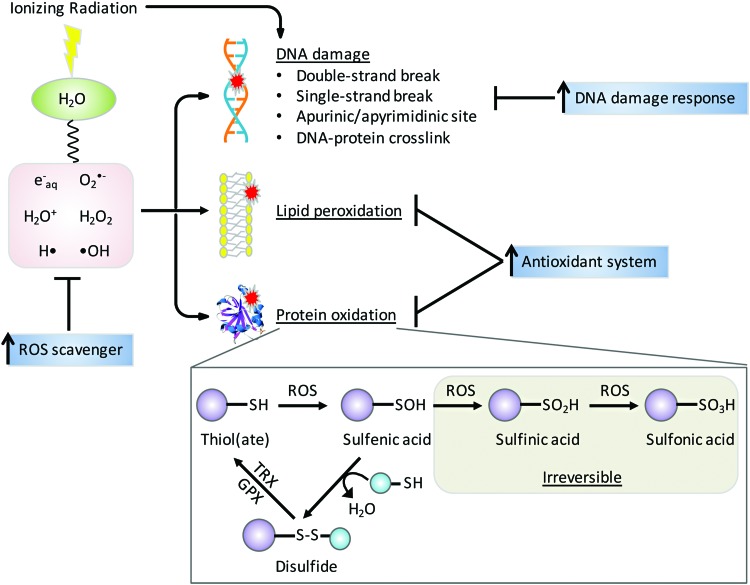
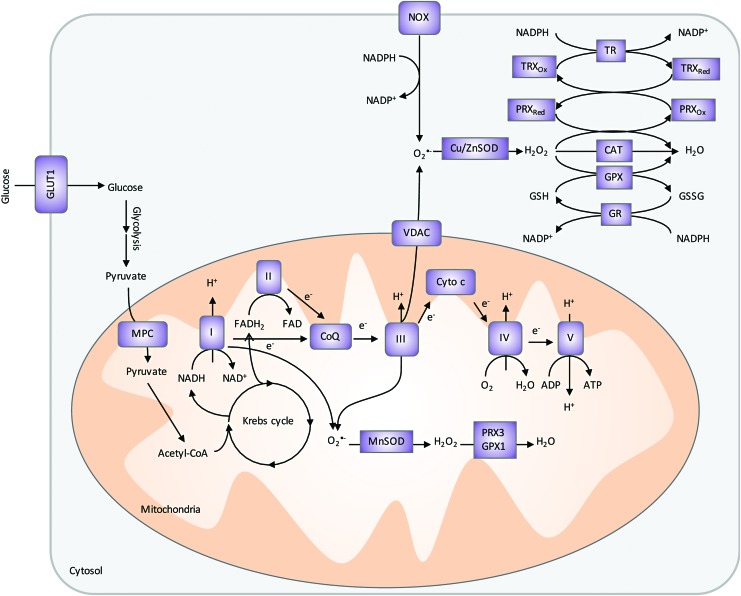
Future directions: As reviewed here, alterations in redox metabolism occur at all stages of HNSCC management, providing opportunities for improved prevention, early detection, response to therapies, and QOL. Bioinformatics and computational systems biology approaches are key to integrate redox effects with multiomics data from cells and clinical specimens and to identify redox modifiers or modifiable target proteins to achieve improved clinical outcomes. Antioxid. Redox Signal.
Keywords: HNSCC; head and neck; oxidative stress; redox; squamous cancer.
Figures
Similar articles
-
Epidermal growth factor receptor targeted therapy in stages III and IV head and neck cancer.Curr Oncol. 2010 Jun;17(3):37-48. doi: 10.3747/co.v17i3.520. Curr Oncol. 2010. PMID: 20567625 Free PMC article.
-
Afatinib versus placebo as adjuvant therapy after chemoradiation in a double-blind, phase III study (LUX-Head & Neck 2) in patients with primary unresected, clinically intermediate-to-high-risk head and neck cancer: study protocol for a randomized controlled trial.Trials. 2014 Nov 29;15:469. doi: 10.1186/1745-6215-15-469. Trials. 2014. PMID: 25432788 Free PMC article. Clinical Trial.
-
Precision medicine in head and neck cancer.Drug Resist Updat. 2018 Sep;40:13-16. doi: 10.1016/j.drup.2018.09.001. Epub 2018 Sep 25. Drug Resist Updat. 2018. PMID: 30466712 Review.
-
SET protein accumulation prevents cell death in head and neck squamous cell carcinoma through regulation of redox state and autophagy.Biochim Biophys Acta Mol Cell Res. 2019 Apr;1866(4):623-637. doi: 10.1016/j.bbamcr.2019.01.005. Epub 2019 Jan 16. Biochim Biophys Acta Mol Cell Res. 2019. PMID: 30658075
-
Evidence-based radiation oncology in head and neck squamous cell carcinoma.Radiother Oncol. 2007 Oct;85(1):156-70. doi: 10.1016/j.radonc.2007.04.002. Epub 2007 May 4. Radiother Oncol. 2007. PMID: 17482300 Review.
Cited by
-
Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue Against Oxidative Injury.Antioxid Redox Signal. 2018 Dec 1;29(16):1691-1724. doi: 10.1089/ars.2017.7453. Epub 2018 Aug 28. Antioxid Redox Signal. 2018. PMID: 29926755 Free PMC article. Review.
-
Initial biological evaluations of 18F-KS1, a novel ascorbate derivative to image oxidative stress in cancer.EJNMMI Res. 2019 May 17;9(1):43. doi: 10.1186/s13550-019-0513-x. EJNMMI Res. 2019. PMID: 31101996 Free PMC article.
-
Redox integration of signaling and metabolism in a head and neck cancer model of radiation resistance using COSMRO.Front Oncol. 2023 Jan 4;12:946320. doi: 10.3389/fonc.2022.946320. eCollection 2022. Front Oncol. 2023. PMID: 36686772 Free PMC article.
-
Establishment and validation of a redox-related long non-coding RNAs prognostic signature in head and neck squamous cell carcinoma.Sci Rep. 2022 Dec 21;12(1):22040. doi: 10.1038/s41598-022-26490-7. Sci Rep. 2022. PMID: 36543836 Free PMC article.
-
Interaction between Cigarette Smoke and Human Papillomavirus 16 E6/E7 Oncoproteins to Induce SOD2 Expression and DNA Damage in Head and Neck Cancer.Int J Mol Sci. 2023 Apr 7;24(8):6907. doi: 10.3390/ijms24086907. Int J Mol Sci. 2023. PMID: 37108069 Free PMC article.
References
-
- Albano E, Tomasi A, Persson J-O, Terelius Y, Goria-Gatti L, Ingelman-Sundberg M, and Dianzani MU. Role of ethanol-inducible cytochrome P450 (P450IIE1) in catalysing the free radical activation of aliphatic alcohols. Biochem Pharmacol 41: 1895–1902, 1991 - PubMed
-
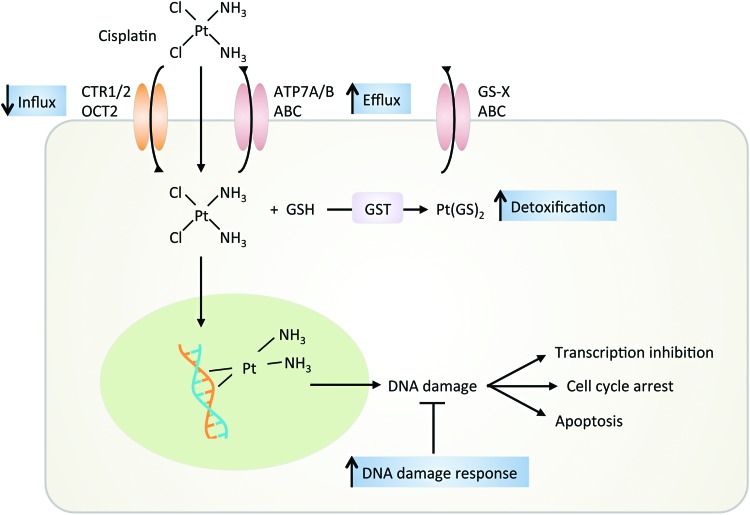
- Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res 106: 27–36, 2016 - PubMed
-
- Anderson CM, Allen BG, Sun W, Agarwala SS, Lee CM, Venigalla ML, Greenberg L, Adkins D, Chen Y, Zhen W, Mould DR, Holmlund J, Brill J, Sonis ST, and Buatti J. Phase 1b trial of superoxide (SO) dismutase (SOD) mimetic GC4419 to reduce chemoradiation therapy (CRT)-induced oral mucositis (OM) in patients (pts) with oral cavity or oropharyngeal carcinoma (OCC). Int J Radiat Oncol Biol Phys 96: S83–S84, 2016 - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
