

Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas
- PMID: 29100075
- PMCID: PMC5693358
- DOI: 10.1016/j.cell.2017.10.014
Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas
Abstract
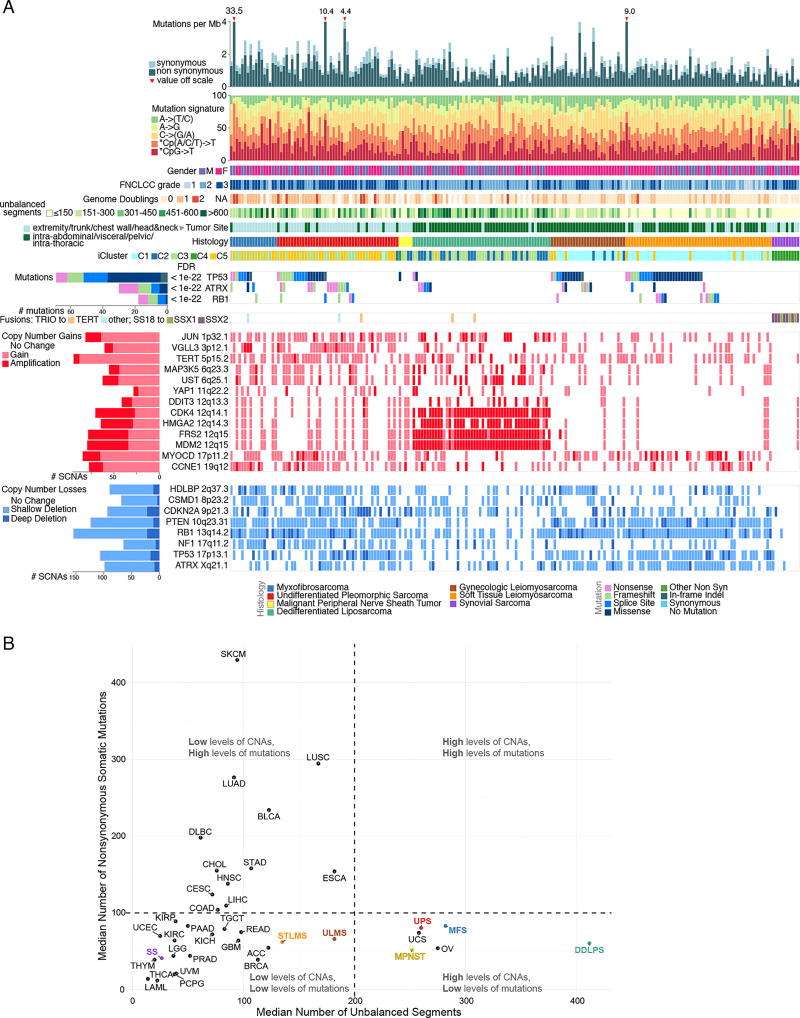
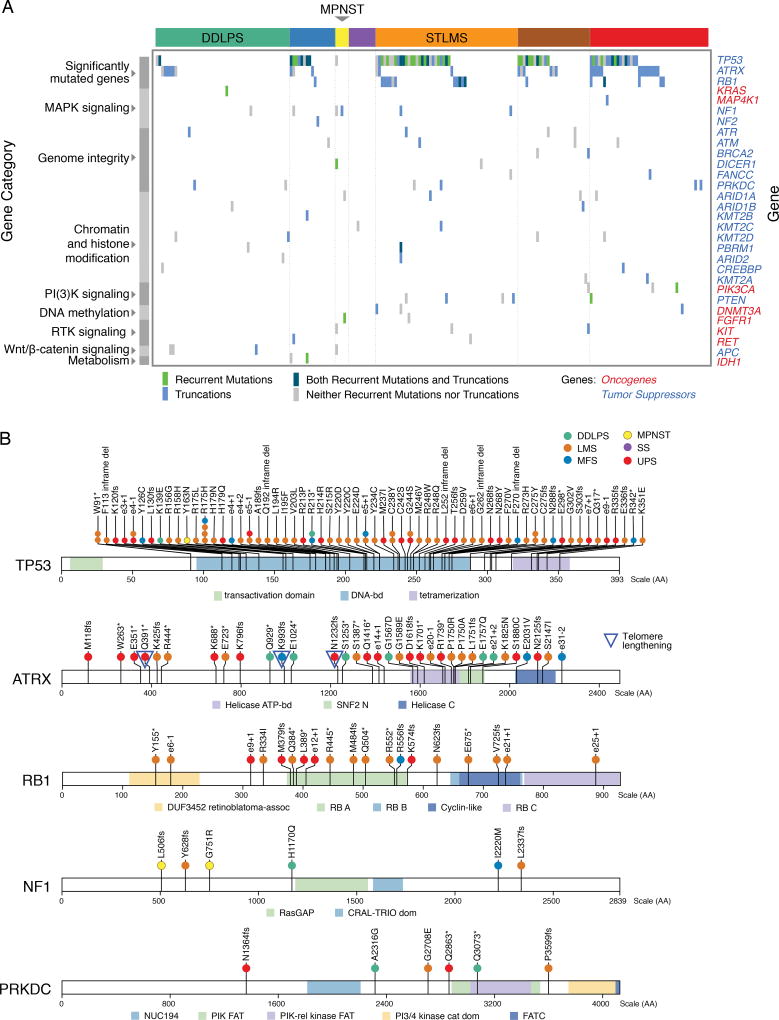
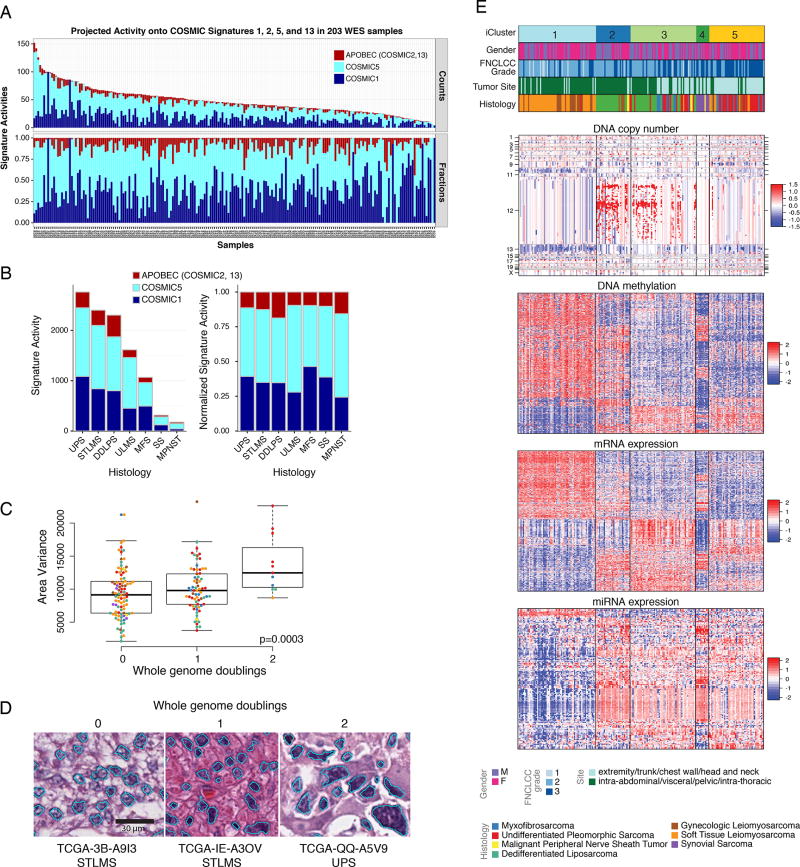
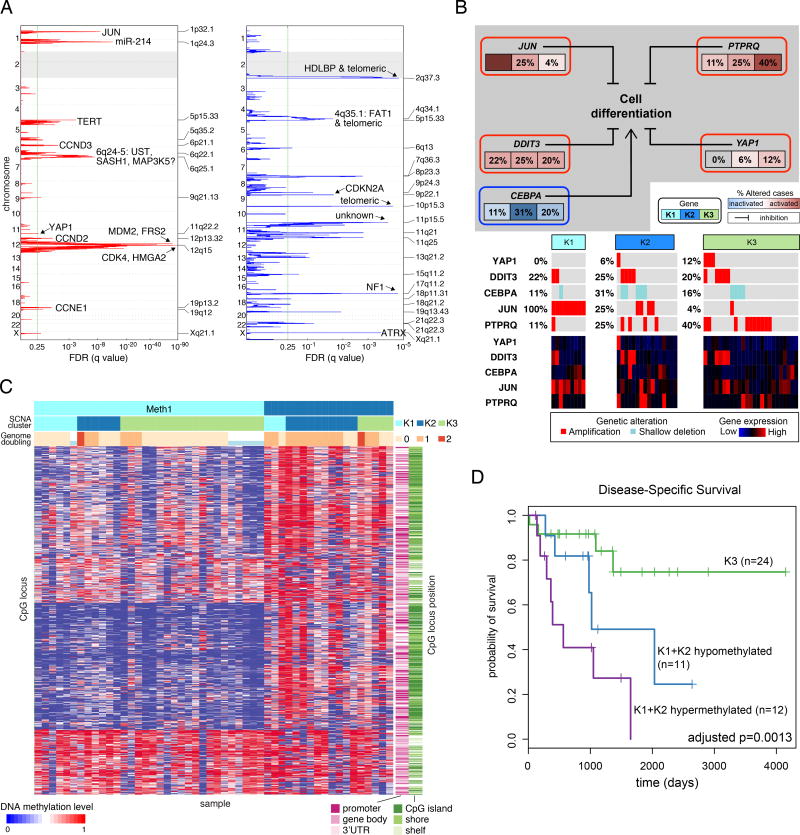
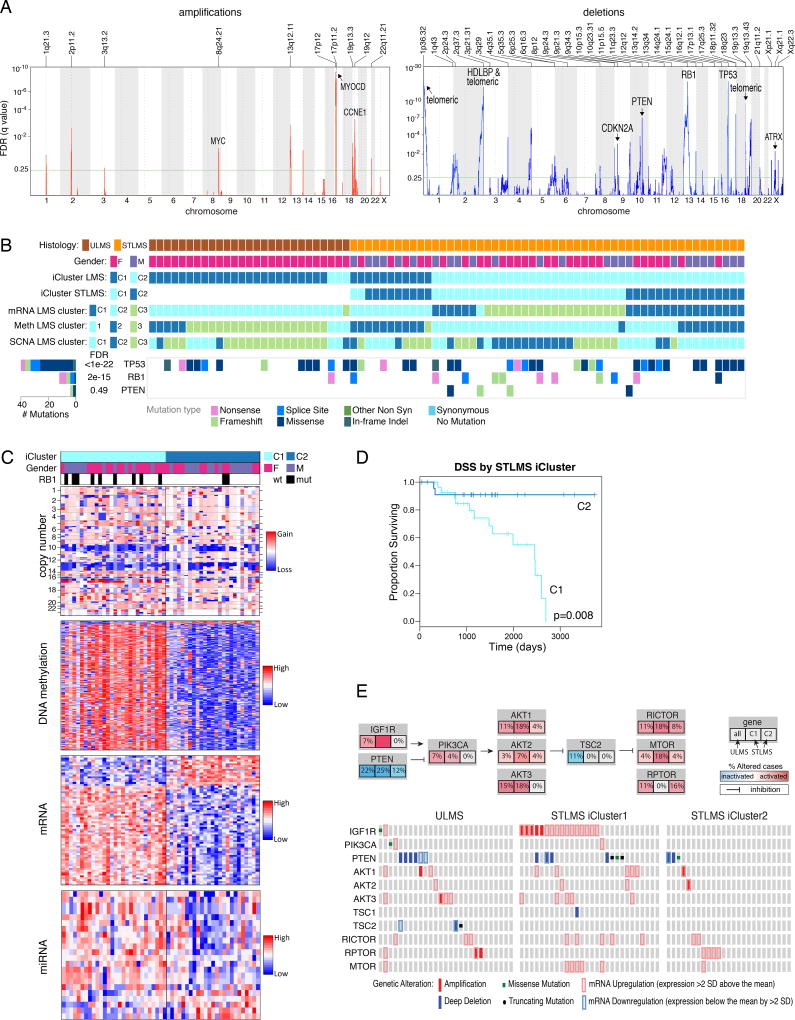
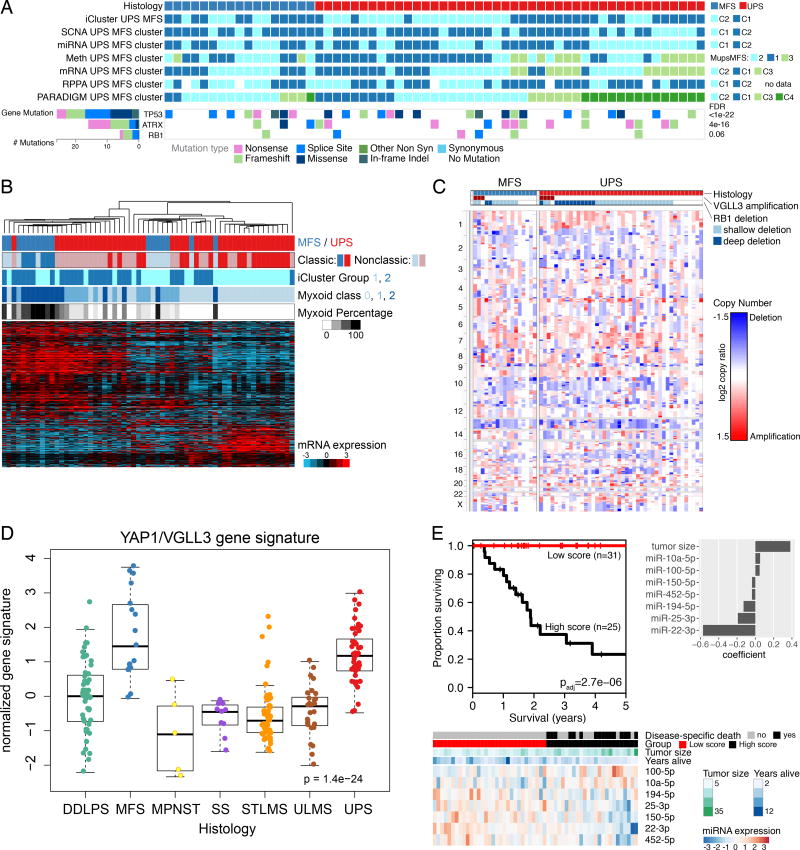
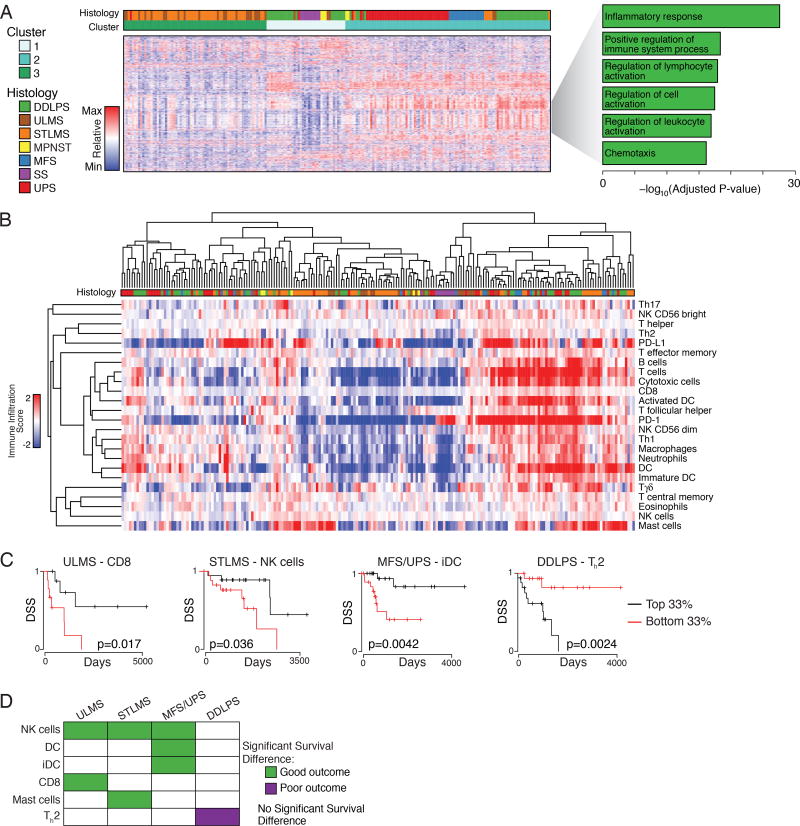
Sarcomas are a broad family of mesenchymal malignancies exhibiting remarkable histologic diversity. We describe the multi-platform molecular landscape of 206 adult soft tissue sarcomas representing 6 major types. Along with novel insights into the biology of individual sarcoma types, we report three overarching findings: (1) unlike most epithelial malignancies, these sarcomas (excepting synovial sarcoma) are characterized predominantly by copy-number changes, with low mutational loads and only a few genes (TP53, ATRX, RB1) highly recurrently mutated across sarcoma types; (2) within sarcoma types, genomic and regulomic diversity of driver pathways defines molecular subtypes associated with patient outcome; and (3) the immune microenvironment, inferred from DNA methylation and mRNA profiles, associates with outcome and may inform clinical trials of immune checkpoint inhibitors. Overall, this large-scale analysis reveals previously unappreciated sarcoma-type-specific changes in copy number, methylation, RNA, and protein, providing insights into refining sarcoma therapy and relationships to other cancer types.
Keywords: DNA methylation; The Cancer Genome Atlas; dedifferentiated liposarcoma; genomics; immune infiltration; leiomyosarcoma; molecular subtype; myxofibrosarcoma; pleomorphism; undifferentiated pleomorphic sarcoma.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas.Cancer. 2017 Sep 1;123(17):3291-3304. doi: 10.1002/cncr.30726. Epub 2017 May 2. Cancer. 2017. PMID: 28463396 Free PMC article.
-
Can DNA Methylation Profiling Classify Histologic Subtypes and Grades in Soft Tissue Sarcoma?Clin Orthop Relat Res. 2024 Mar 22;482(6):1028-37. doi: 10.1097/CORR.0000000000003041. Online ahead of print. Clin Orthop Relat Res. 2024. PMID: 38517415
-
Integration of genomic copy number variations and chemotherapy-response biomarkers in pediatric sarcoma.BMC Med Genomics. 2019 Jan 31;12(Suppl 1):23. doi: 10.1186/s12920-018-0456-5. BMC Med Genomics. 2019. PMID: 30704460 Free PMC article.
-
Immunotherapy for sarcomas.Jpn J Clin Oncol. 2021 Apr 1;51(4):523-537. doi: 10.1093/jjco/hyab005. Jpn J Clin Oncol. 2021. PMID: 33611603 Review.
-
Subclassification of pleomorphic sarcomas: How and why should we care?Ann Diagn Pathol. 2018 Dec;37:118-124. doi: 10.1016/j.anndiagpath.2018.10.006. Epub 2018 Oct 11. Ann Diagn Pathol. 2018. PMID: 30340082 Review.
Cited by
-
Novel Nomograms-Based Prediction Models for Patients with Primary Undifferentiated Pleomorphic Sarcomas Resections.Cancers (Basel). 2021 Apr 15;13(8):1917. doi: 10.3390/cancers13081917. Cancers (Basel). 2021. PMID: 33921187 Free PMC article.
-
Differential quantities of immune checkpoint-expressing CD8 T cells in soft tissue sarcoma subtypes.J Immunother Cancer. 2020 Aug;8(2):e000271. doi: 10.1136/jitc-2019-000271. J Immunother Cancer. 2020. PMID: 32792357 Free PMC article.
-
Cellular origin and clonal evolution of human dedifferentiated liposarcoma.Nat Commun. 2024 Sep 12;15(1):7941. doi: 10.1038/s41467-024-52067-1. Nat Commun. 2024. PMID: 39266532 Free PMC article.
-
Core transcriptional regulatory circuitries in cancer.Oncogene. 2020 Oct;39(43):6633-6646. doi: 10.1038/s41388-020-01459-w. Epub 2020 Sep 17. Oncogene. 2020. PMID: 32943730 Free PMC article. Review.
-
Virus-Driven Carcinogenesis.Cancers (Basel). 2021 May 27;13(11):2625. doi: 10.3390/cancers13112625. Cancers (Basel). 2021. PMID: 34071792 Free PMC article. Review.
References
-
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. - PubMed
MeSH terms
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA143882/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA210974/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA126543/CA/NCI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- R50 CA221675/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA194362/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA210999/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA199461/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
