

CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy
- PMID: 29078326
- PMCID: PMC5676903
- DOI: 10.1073/pnas.1708268114
CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy
Abstract
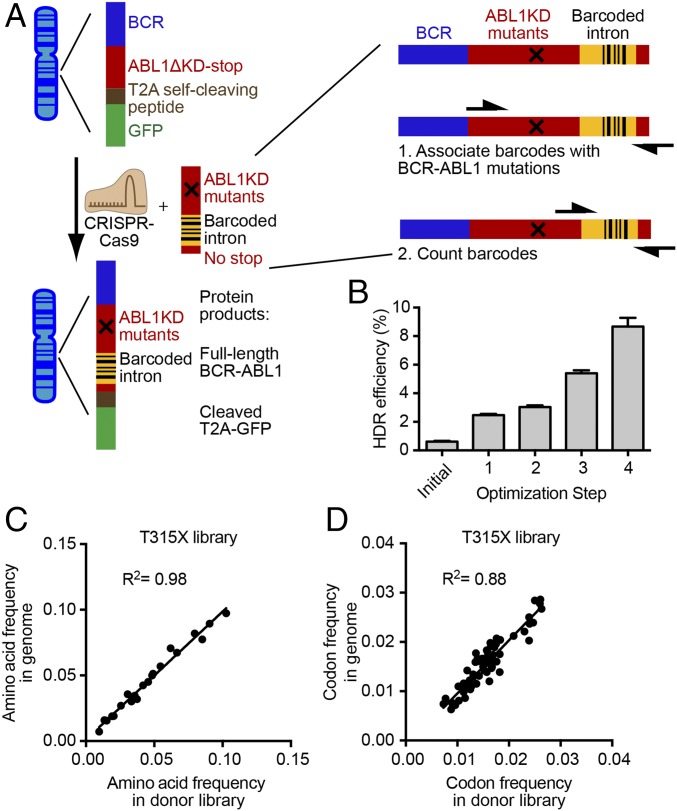
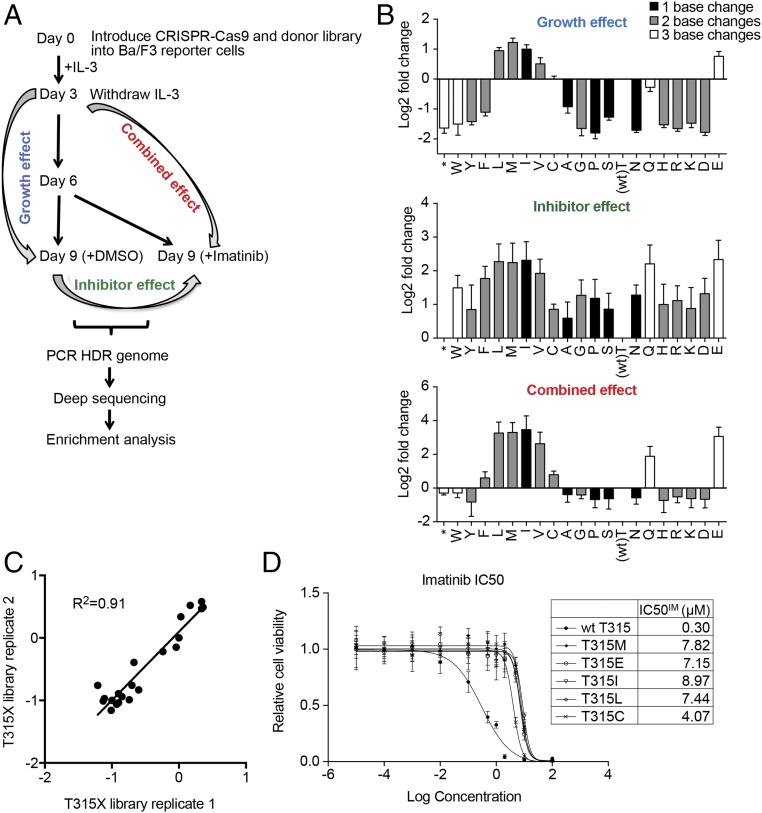
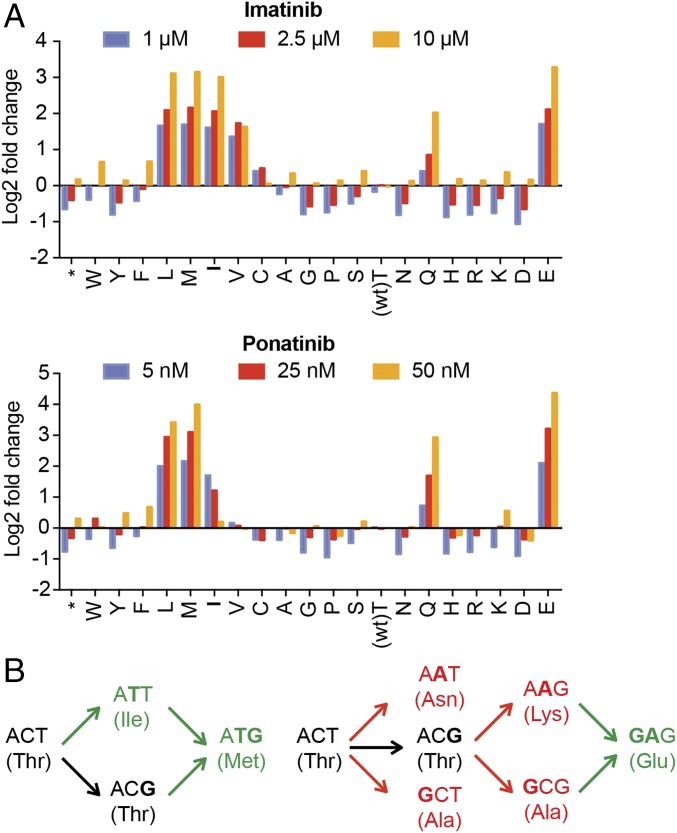
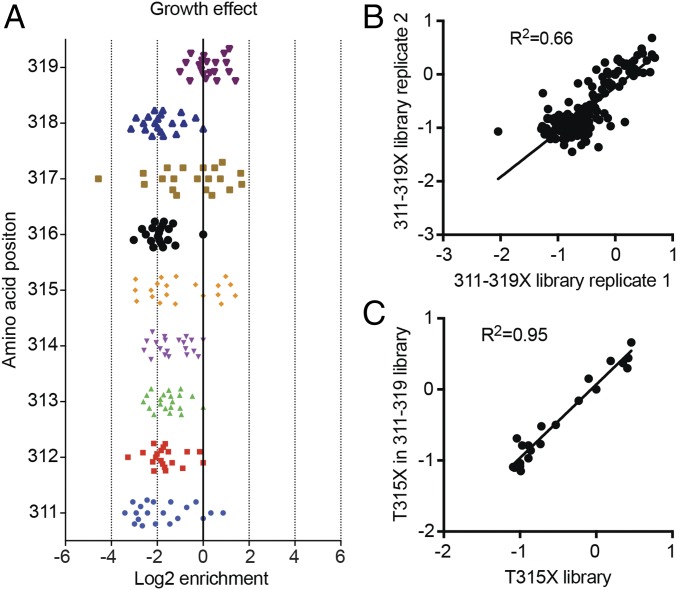
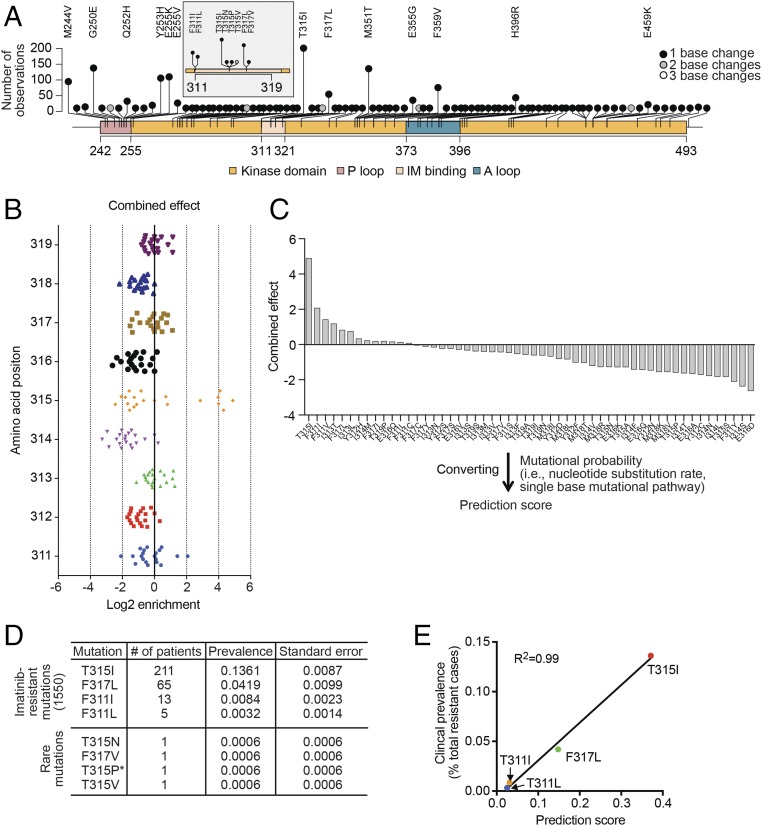
Developing tools to accurately predict the clinical prevalence of drug-resistant mutations is a key step toward generating more effective therapeutics. Here we describe a high-throughput CRISPR-Cas9-based saturated mutagenesis approach to generate comprehensive libraries of point mutations at a defined genomic location and systematically study their effect on cell growth. As proof of concept, we mutagenized a selected region within the leukemic oncogene BCR-ABL1 Using bulk competitions with a deep-sequencing readout, we analyzed hundreds of mutations under multiple drug conditions and found that the effects of mutations on growth in the presence or absence of drug were critical for predicting clinically relevant resistant mutations, many of which were cancer adaptive in the absence of drug pressure. Using this approach, we identified all clinically isolated BCR-ABL1 mutations and achieved a prediction score that correlated highly with their clinical prevalence. The strategy described here can be broadly applied to a variety of oncogenes to predict patient mutations and evaluate resistance susceptibility in the development of new therapeutics.
Keywords: BCR-ABL; CRISPR-Cas9–based genome editing; drug resistance; saturated mutagenesis; tyrosine kinase inhibitors.
Published under the PNAS license.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Use of CRISPR/Cas Genome Editing Technology for Targeted Mutagenesis in Rice.Methods Mol Biol. 2017;1498:33-40. doi: 10.1007/978-1-4939-6472-7_3. Methods Mol Biol. 2017. PMID: 27709567
-
Characteristic and inheritance analysis of targeted mutagenesis mediated by genome editing in rice.Yi Chuan. 2016 Aug;38(8):746-55. doi: 10.16288/j.yczz.16-052. Yi Chuan. 2016. PMID: 27531613
-
Generation of lung cancer cell lines harboring EGFR T790M mutation by CRISPR/Cas9-mediated genome editing.Oncotarget. 2017 May 30;8(22):36331-36338. doi: 10.18632/oncotarget.16752. Oncotarget. 2017. PMID: 28422737 Free PMC article.
-
The CRISPR/Cas9 system: Their delivery, in vivo and ex vivo applications and clinical development by startups.Biotechnol Prog. 2017 Jul;33(4):1035-1045. doi: 10.1002/btpr.2484. Epub 2017 May 14. Biotechnol Prog. 2017. PMID: 28440027 Review.
-
Advances in therapeutic CRISPR/Cas9 genome editing.Transl Res. 2016 Feb;168:15-21. doi: 10.1016/j.trsl.2015.09.008. Epub 2015 Sep 26. Transl Res. 2016. PMID: 26470680 Review.
Cited by
-
The Pseudomonas aeruginosa Resistome: Permanent and Transient Antibiotic Resistance, an Overview.Methods Mol Biol. 2024;2721:85-102. doi: 10.1007/978-1-0716-3473-8_7. Methods Mol Biol. 2024. PMID: 37819517
-
From systems to structure - using genetic data to model protein structures.Nat Rev Genet. 2022 Jun;23(6):342-354. doi: 10.1038/s41576-021-00441-w. Epub 2022 Jan 10. Nat Rev Genet. 2022. PMID: 35013567 Free PMC article. Review.
-
Saturation mutagenesis of a predicted ancestral Syk-family kinase.Protein Sci. 2022 Oct;31(10):e4411. doi: 10.1002/pro.4411. Protein Sci. 2022. PMID: 36173161 Free PMC article.
-
Understanding molecular mechanisms in cell signaling through natural and artificial sequence variation.Nat Struct Mol Biol. 2019 Jan;26(1):25-34. doi: 10.1038/s41594-018-0175-9. Epub 2018 Dec 31. Nat Struct Mol Biol. 2019. PMID: 30598552 Free PMC article. Review.
-
Applications of Protein Engineering and Directed Evolution in Plant Research.Plant Physiol. 2019 Mar;179(3):907-917. doi: 10.1104/pp.18.01534. Epub 2019 Jan 9. Plant Physiol. 2019. PMID: 30626612 Free PMC article. Review.
References
-
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. - PubMed
-
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: An evolving paradigm. Nat Rev Cancer. 2013;13:714–726. - PubMed
-
- Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. - PubMed
-
- Ma Y, et al. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods. 2016;13:1029–1035. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
