

Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides
- PMID: 29078297
- PMCID: PMC5692539
- DOI: 10.1073/pnas.1706489114
Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides
Erratum in
-
Correction for Kang et al., Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides.Proc Natl Acad Sci U S A. 2017 Dec 19;114(51):E11057. doi: 10.1073/pnas.1719887115. Epub 2017 Nov 27. Proc Natl Acad Sci U S A. 2017. PMID: 29180413 Free PMC article. No abstract available.
Abstract
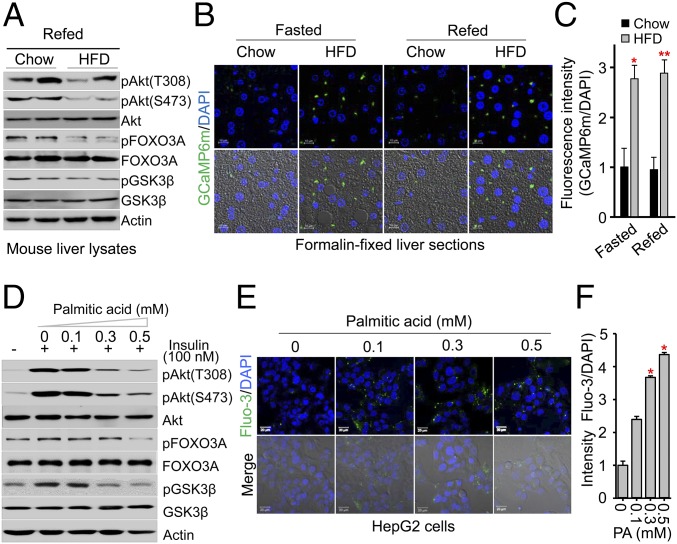
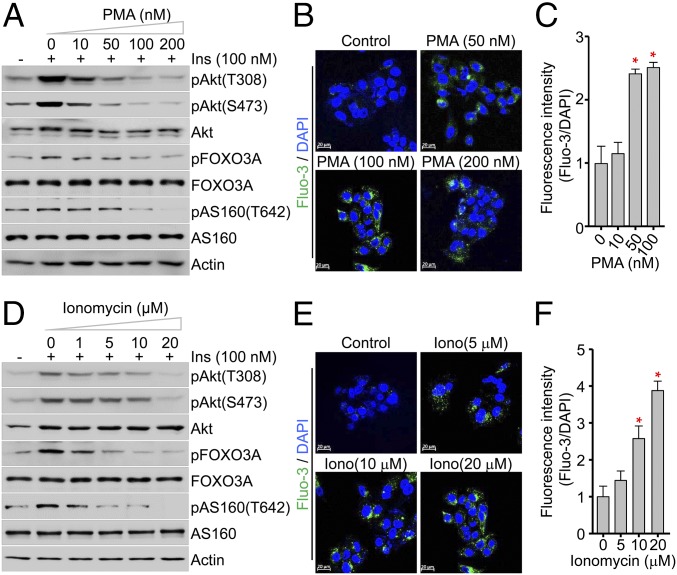
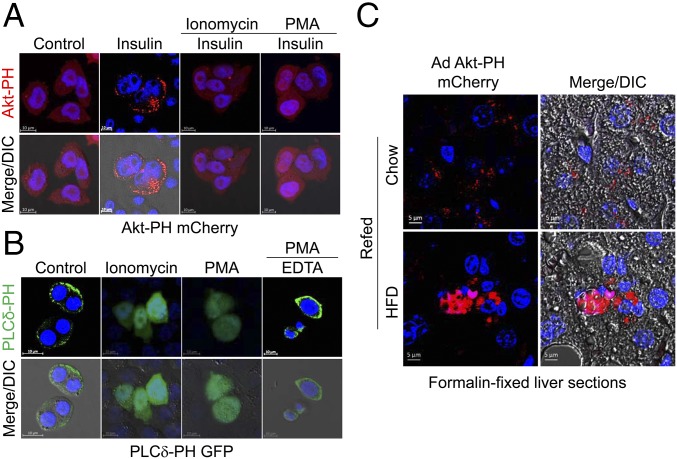
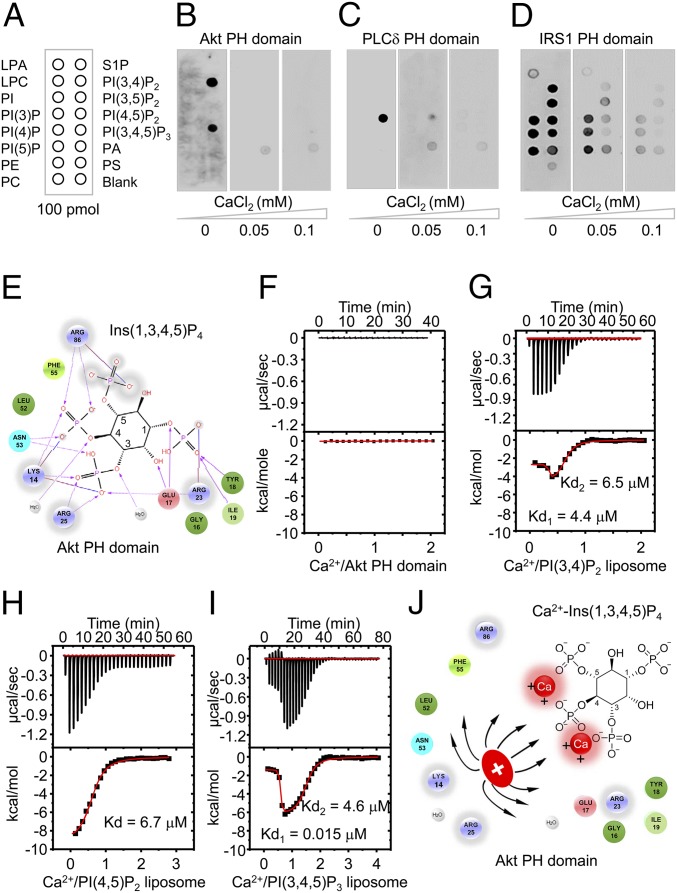
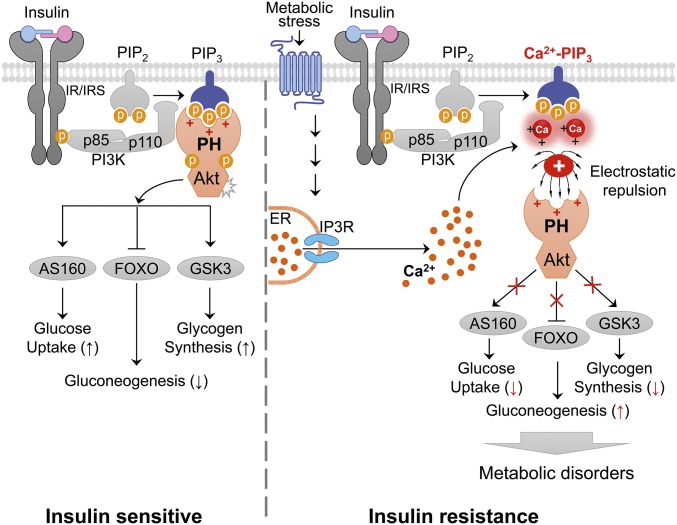
Insulin resistance, a key etiological factor in metabolic syndrome, is closely linked to ectopic lipid accumulation and increased intracellular Ca2+ concentrations in muscle and liver. However, the mechanism by which dysregulated intracellular Ca2+ homeostasis causes insulin resistance remains elusive. Here, we show that increased intracellular Ca2+ acts as a negative regulator of insulin signaling. Chronic intracellular Ca2+ overload in hepatocytes during obesity and hyperlipidemia attenuates the phosphorylation of protein kinase B (Akt) and its key downstream signaling molecules by inhibiting membrane localization of pleckstrin homology (PH) domains. Pharmacological approaches showed that elevated intracellular Ca2+ inhibits insulin-stimulated Akt phosphorylation and abrogates membrane localization of various PH domain proteins such as phospholipase Cδ and insulin receptor substrate 1, suggesting a common mechanism inhibiting the membrane targeting of PH domains. PH domain-lipid overlay assays confirmed that Ca2+ abolishes the binding of various PH domains to phosphoinositides (PIPs) with two adjacent phosphate groups, such as PI(3,4)P2, PI(4,5)P2, and PI(3,4,5)P3 Finally, thermodynamic analysis of the binding interaction showed that Ca2+-mediated inhibition of targeting PH domains to the membrane resulted from the tight binding of Ca2+ rather than PH domains to PIPs forming Ca2+-PIPs. Thus, Ca2+-PIPs prevent the recognition of PIPs by PH domains, potentially due to electrostatic repulsion between positively charged side chains in PH domains and the Ca2+-PIPs. Our findings provide a mechanistic link between intracellular Ca2+ dysregulation and Akt inactivation in insulin resistance.
Keywords: Ca2+-phosphoinositides; PH domain; insulin resistance; intracellular Ca2+ concentration; membrane localization.
Copyright © 2017 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Phosphoinositides and intracellular calcium signaling: novel insights into phosphoinositides and calcium coupling as negative regulators of cellular signaling.Exp Mol Med. 2023 Aug;55(8):1702-1712. doi: 10.1038/s12276-023-01067-0. Epub 2023 Aug 1. Exp Mol Med. 2023. PMID: 37524877 Free PMC article. Review.
-
Candesartan, an angiotensin-II receptor blocker, ameliorates insulin resistance and hepatosteatosis by reducing intracellular calcium overload and lipid accumulation.Exp Mol Med. 2023 May;55(5):910-925. doi: 10.1038/s12276-023-00982-6. Epub 2023 May 1. Exp Mol Med. 2023. PMID: 37121975 Free PMC article.
-
Different subcellular localization and phosphoinositides binding of insulin receptor substrate protein pleckstrin homology domains.Mol Endocrinol. 2000 Jun;14(6):823-36. doi: 10.1210/mend.14.6.0486. Mol Endocrinol. 2000. PMID: 10847585
-
TRAF6-mediated ubiquitination of APPL1 enhances hepatic actions of insulin by promoting the membrane translocation of Akt.Biochem J. 2013 Oct 15;455(2):207-16. doi: 10.1042/BJ20130760. Biochem J. 2013. PMID: 23909487
-
Cellular and molecular interactions of phosphoinositides and peripheral proteins.Chem Phys Lipids. 2014 Sep;182:3-18. doi: 10.1016/j.chemphyslip.2014.02.002. Epub 2014 Feb 17. Chem Phys Lipids. 2014. PMID: 24556335 Free PMC article. Review.
Cited by
-
Equivalent change enrichment analysis: assessing equivalent and inverse change in biological pathways between diverse experiments.BMC Genomics. 2020 Feb 24;21(1):180. doi: 10.1186/s12864-020-6589-x. BMC Genomics. 2020. PMID: 32093613 Free PMC article.
-
Chitosan-Selenium Nanoparticle (Cs-Se NP) Foliar Spray Alleviates Salt Stress in Bitter Melon.Nanomaterials (Basel). 2021 Mar 9;11(3):684. doi: 10.3390/nano11030684. Nanomaterials (Basel). 2021. PMID: 33803416 Free PMC article.
-
High-phytate/low-calcium diet is a risk factor for crystal nephropathies, renal phosphate wasting, and bone loss.Elife. 2020 Apr 9;9:e52709. doi: 10.7554/eLife.52709. Elife. 2020. PMID: 32271147 Free PMC article.
-
High-Phytate Diets Increase Amyloid β Deposition and Apoptotic Neuronal Cell Death in a Rat Model.Nutrients. 2021 Dec 6;13(12):4370. doi: 10.3390/nu13124370. Nutrients. 2021. PMID: 34959925 Free PMC article.
-
The engagement of autophagy in maniac disease.CNS Neurosci Ther. 2023 Dec;29(12):3684-3692. doi: 10.1111/cns.14353. Epub 2023 Jul 12. CNS Neurosci Ther. 2023. PMID: 37438945 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
