

Autophagy inhibitor 3-methyladenine protects against endothelial cell barrier dysfunction in acute lung injury
- PMID: 29074492
- PMCID: PMC5900361
- DOI: 10.1152/ajplung.00555.2016
Autophagy inhibitor 3-methyladenine protects against endothelial cell barrier dysfunction in acute lung injury
Abstract
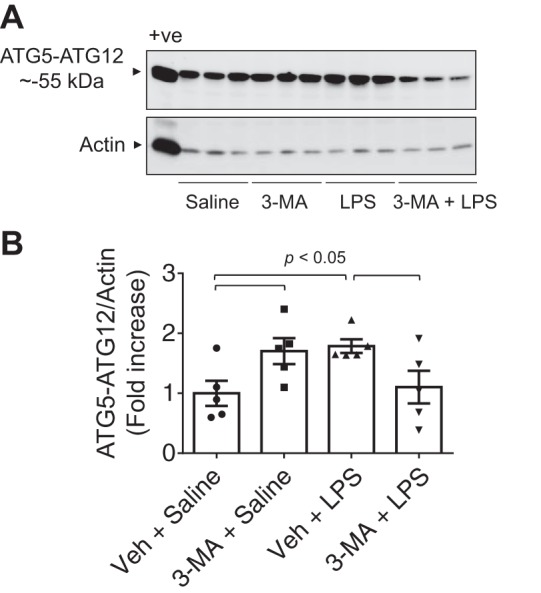
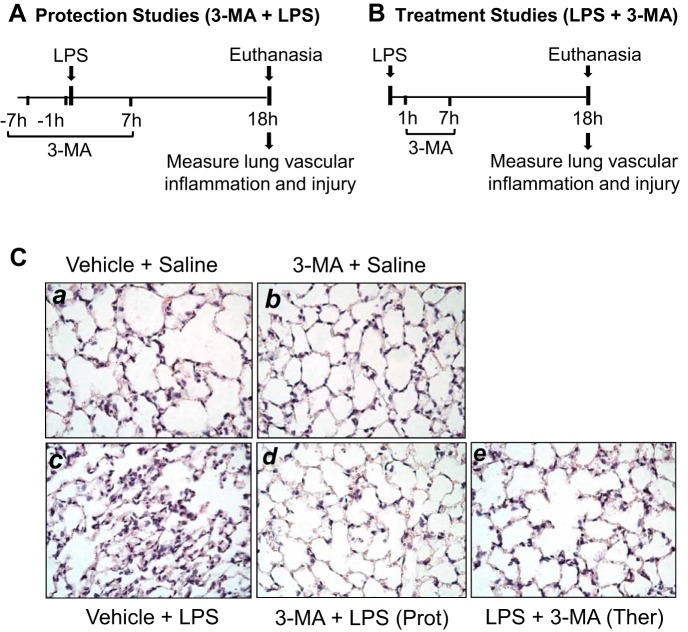
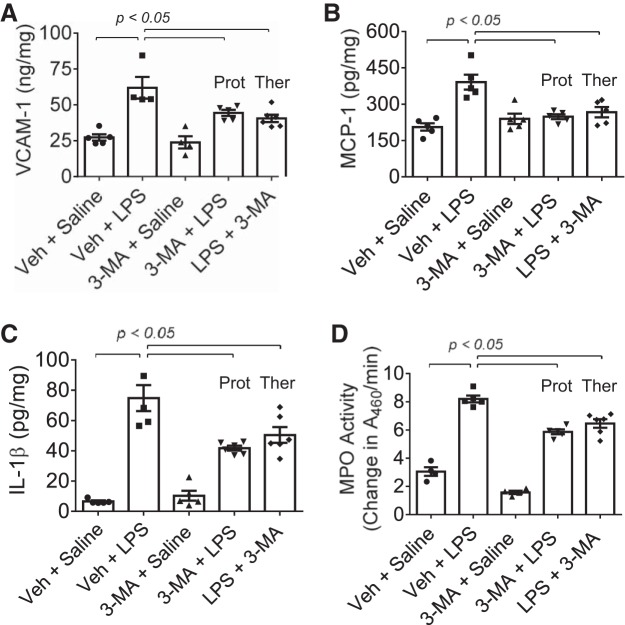
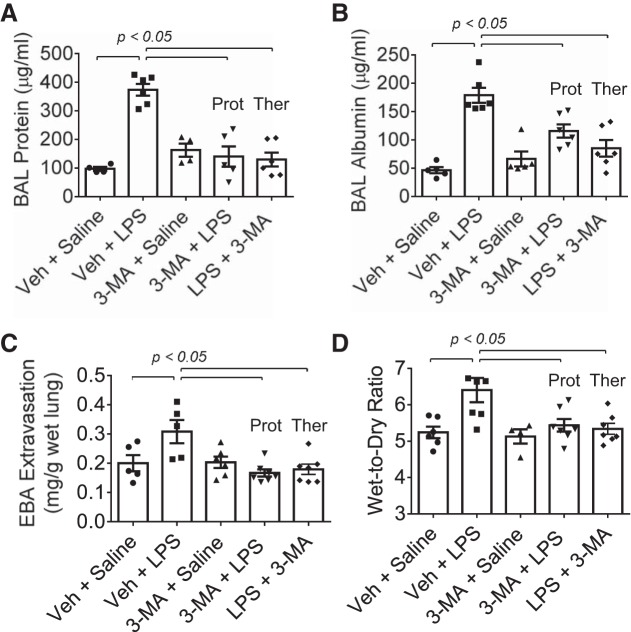
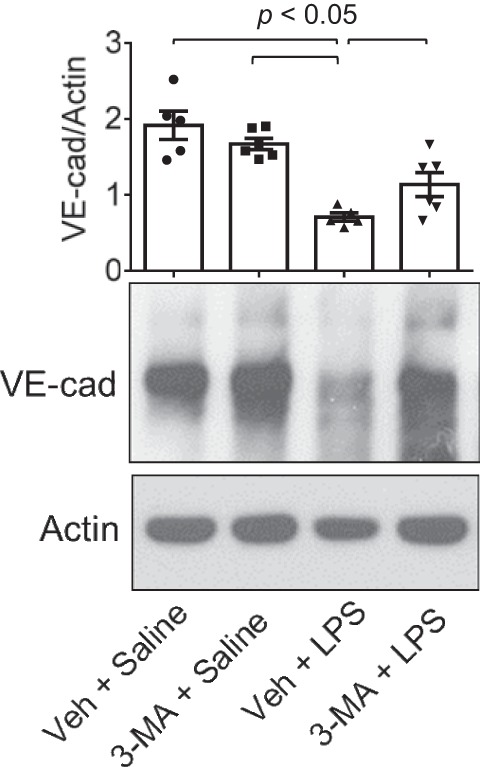
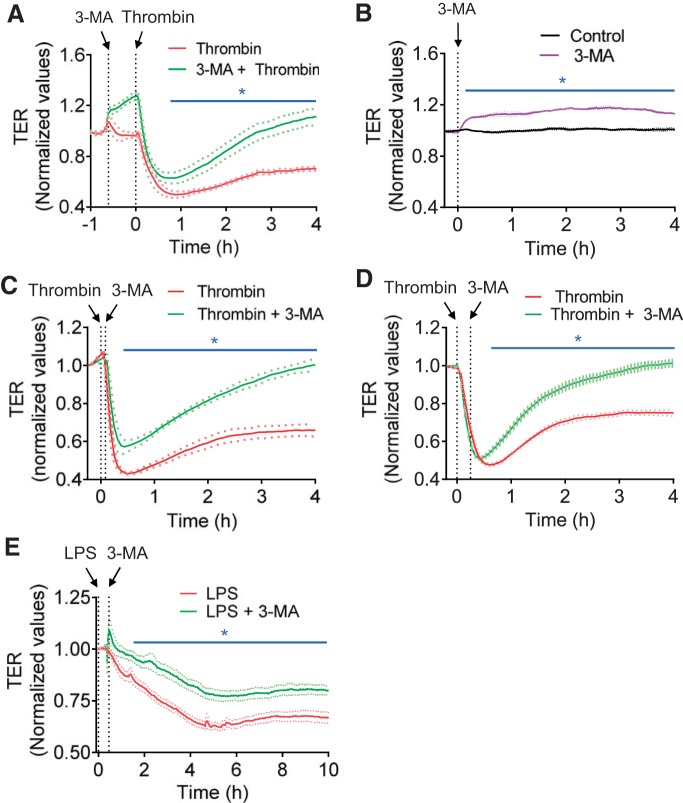
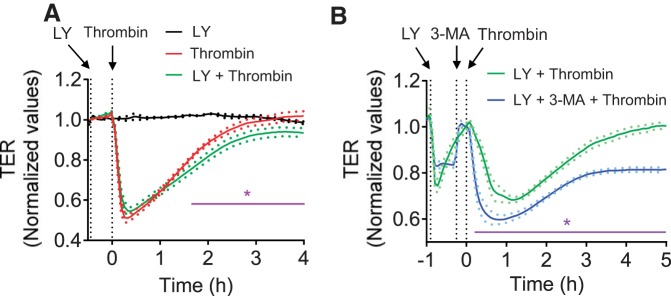
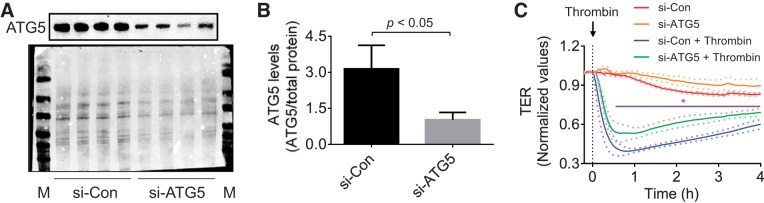
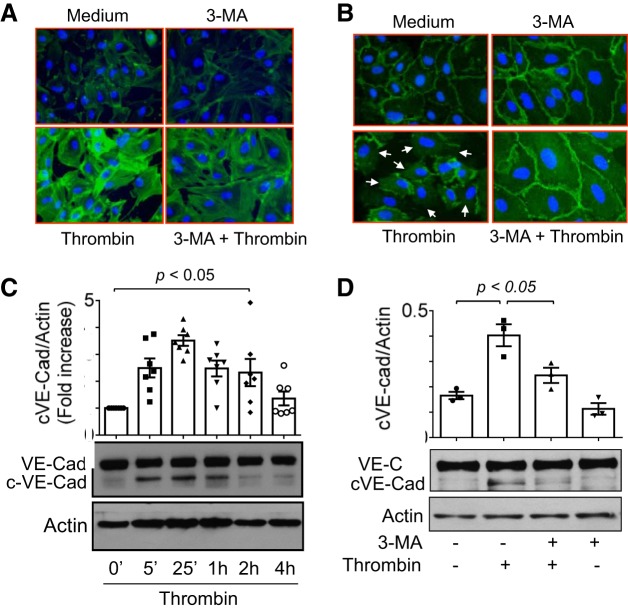
Autophagy is an evolutionarily conserved cellular process that facilitates the continuous recycling of intracellular components (organelles and proteins) and provides an alternative source of energy when nutrients are scarce. Recent studies have implicated autophagy in many disorders, including pulmonary diseases. However, the role of autophagy in endothelial cell (EC) barrier dysfunction and its relevance in the context of acute lung injury (ALI) remain uncertain. Here, we provide evidence that autophagy is a critical component of EC barrier disruption in ALI. Using an aerosolized bacterial lipopolysaccharide (LPS) inhalation mouse model of ALI, we found that administration of the autophagy inhibitor 3-methyladenine (3-MA), either prophylactically or therapeutically, markedly reduced lung vascular leakage and tissue edema. 3-MA was also effective in reducing the levels of proinflammatory mediators and lung neutrophil sequestration induced by LPS. To test the possibility that autophagy in EC could contribute to lung vascular injury, we addressed its role in the mechanism of EC barrier disruption. Knockdown of ATG5, an essential regulator of autophagy, attenuated thrombin-induced EC barrier disruption, confirming the involvement of autophagy in the response. Similarly, exposure of cells to 3-MA, either before or after thrombin, protected against EC barrier dysfunction by inhibiting the cleavage and loss of vascular endothelial cadherin at adherens junctions, as well as formation of actin stress fibers. 3-MA also reversed LPS-induced EC barrier disruption. Together, these data imply a role of autophagy in lung vascular injury and reveal the protective and therapeutic utility of 3-MA against ALI.
Keywords: adherens junctions; autophagy; endothelial cells; lung vascular injury.
Figures
Similar articles
-
Phospholipase C-ε signaling mediates endothelial cell inflammation and barrier disruption in acute lung injury.Am J Physiol Lung Cell Mol Physiol. 2016 Aug 1;311(2):L517-24. doi: 10.1152/ajplung.00069.2016. Epub 2016 Jul 1. Am J Physiol Lung Cell Mol Physiol. 2016. PMID: 27371732 Free PMC article.
-
RAB26-dependent autophagy protects adherens junctional integrity in acute lung injury.Autophagy. 2018;14(10):1677-1692. doi: 10.1080/15548627.2018.1476811. Epub 2018 Jul 26. Autophagy. 2018. PMID: 29965781 Free PMC article.
-
p18, a novel adaptor protein, regulates pulmonary endothelial barrier function via enhanced endocytic recycling of VE-cadherin.FASEB J. 2015 Mar;29(3):868-81. doi: 10.1096/fj.14-257212. Epub 2014 Nov 17. FASEB J. 2015. PMID: 25404710 Free PMC article.
-
Oxidized Phospholipids in Control of Endothelial Barrier Function: Mechanisms and Implication in Lung Injury.Front Endocrinol (Lausanne). 2021 Nov 23;12:794437. doi: 10.3389/fendo.2021.794437. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 34887839 Free PMC article. Review.
-
Direct effects of endotoxin on the endothelium: barrier function and injury.Lab Invest. 1999 Oct;79(10):1181-99. Lab Invest. 1999. PMID: 10532583 Review.
Cited by
-
Phosphodiesterase 10A Is a Key Mediator of Lung Inflammation.J Immunol. 2021 Jun 15;206(12):3010-3020. doi: 10.4049/jimmunol.2001026. Epub 2021 Jun 11. J Immunol. 2021. PMID: 34117108 Free PMC article.
-
Based on Bioinformatics to Explore the Mechanism of "Tangzhiqing" Decoction Alleviating Type 2 Diabetes-associated Cognitive Dysfunction in Mice by Regulating Hippocampal Neuron Apoptosis and Autophagy.Comb Chem High Throughput Screen. 2024;27(17):2565-2582. doi: 10.2174/0113862073255849231030114405. Comb Chem High Throughput Screen. 2024. PMID: 37990900
-
Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases.Cells. 2019 Feb 3;8(2):123. doi: 10.3390/cells8020123. Cells. 2019. PMID: 30717487 Free PMC article. Review.
-
Autophagy, Unfolded Protein Response and Lung Disease.Curr Res Cell Biol. 2020;1:100003. doi: 10.1016/j.crcbio.2020.100003. Epub 2020 Oct 15. Curr Res Cell Biol. 2020. PMID: 33163960 Free PMC article.
-
TM9SF1 knockdown decreases inflammation by enhancing autophagy in a mouse model of acute lung injury.Heliyon. 2022 Dec 5;8(12):e12092. doi: 10.1016/j.heliyon.2022.e12092. eCollection 2022 Dec. Heliyon. 2022. PMID: 36561687 Free PMC article.
References
-
- Bijli KM, Fazal F, Slavin SA, Leonard A, Grose V, Alexander WB, Smrcka AV, Rahman A. Phospholipase C-ε signaling mediates endothelial cell inflammation and barrier disruption in acute lung injury. Am J Physiol Lung Cell Mol Physiol 311: L517–L524, 2016. doi:10.1152/ajplung.00069.2016. - DOI - PMC - PubMed
-
- Brest P, Corcelle EA, Cesaro A, Chargui A, Belaïd A, Klionsky DJ, Vouret-Craviari V, Hebuterne X, Hofman P, Mograbi B. Autophagy and Crohn’s disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr Mol Med 10: 486–502, 2010. doi:10.2174/156652410791608252. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources