

Absence of Carbohydrate Response Element Binding Protein in Adipocytes Causes Systemic Insulin Resistance and Impairs Glucose Transport
- PMID: 29069585
- PMCID: PMC5771491
- DOI: 10.1016/j.celrep.2017.09.091
Absence of Carbohydrate Response Element Binding Protein in Adipocytes Causes Systemic Insulin Resistance and Impairs Glucose Transport
Abstract
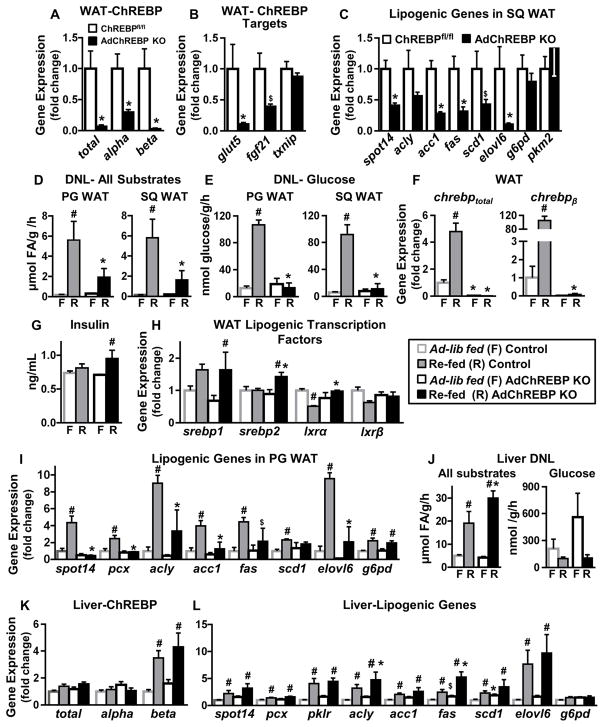
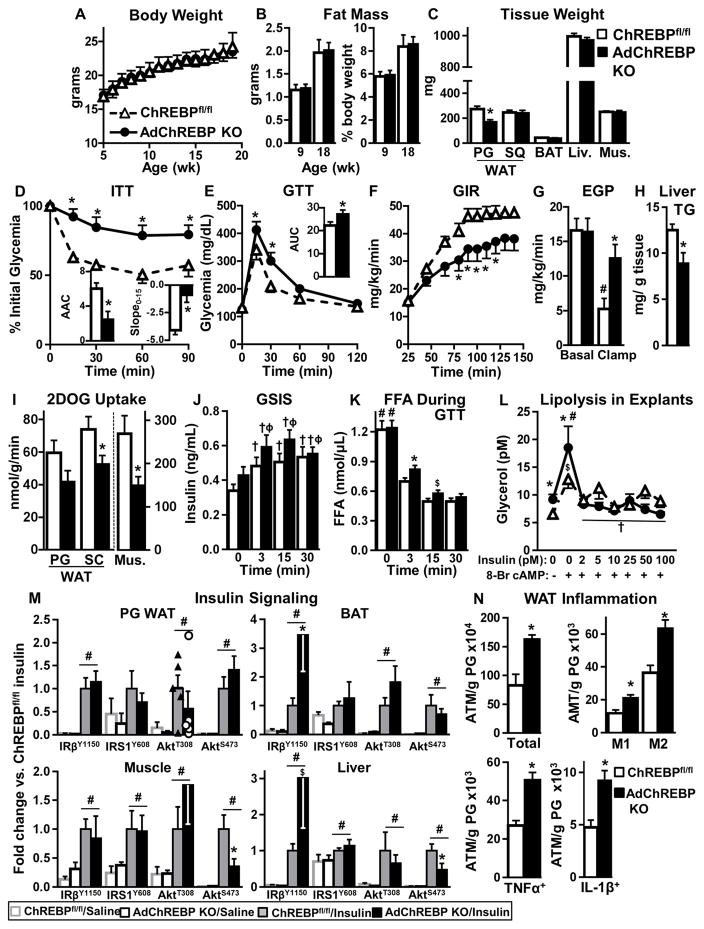
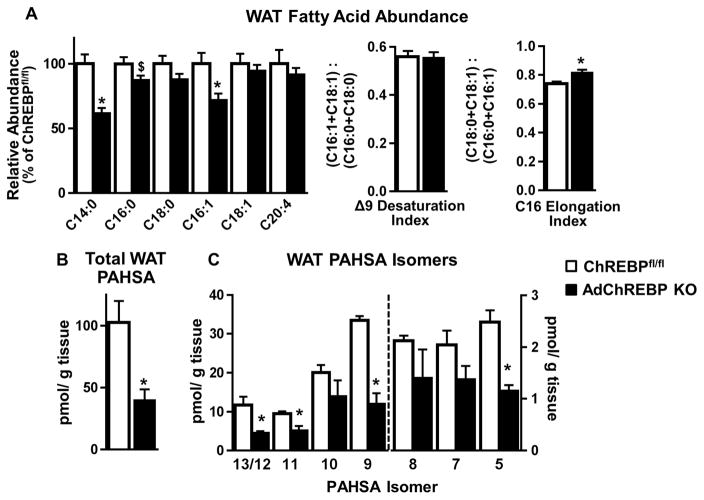
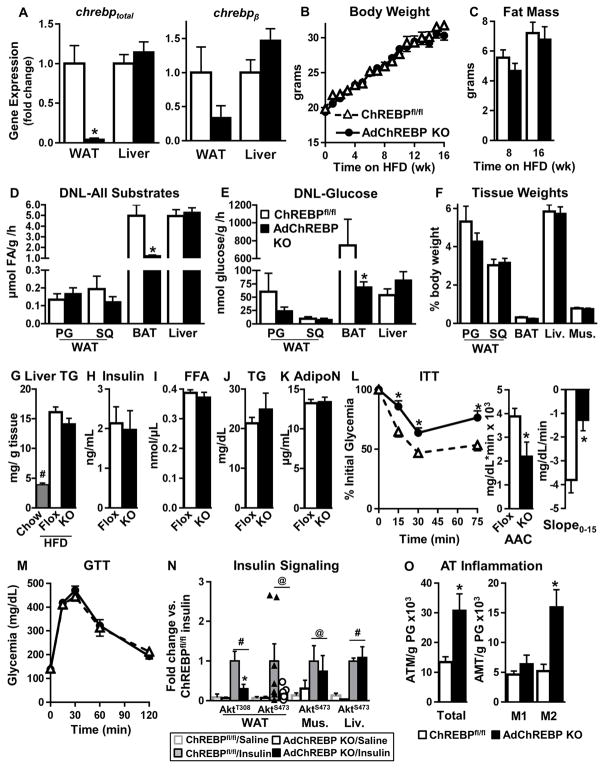
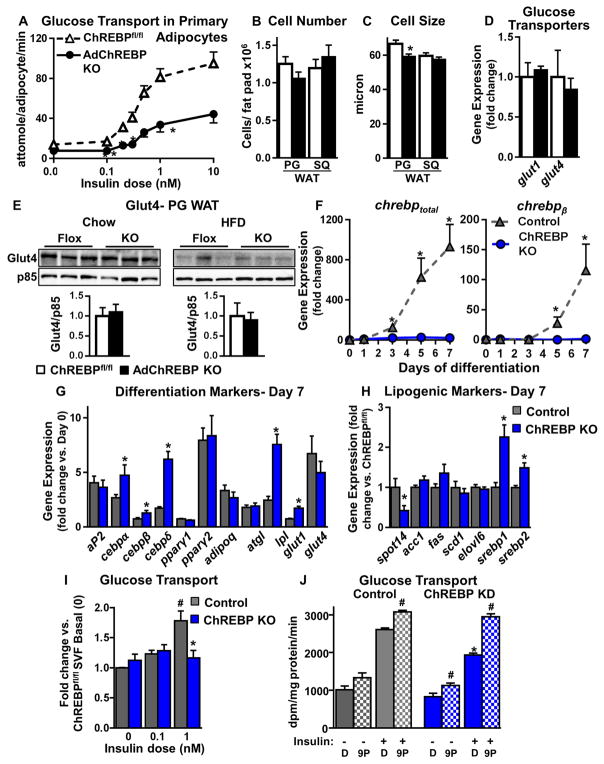
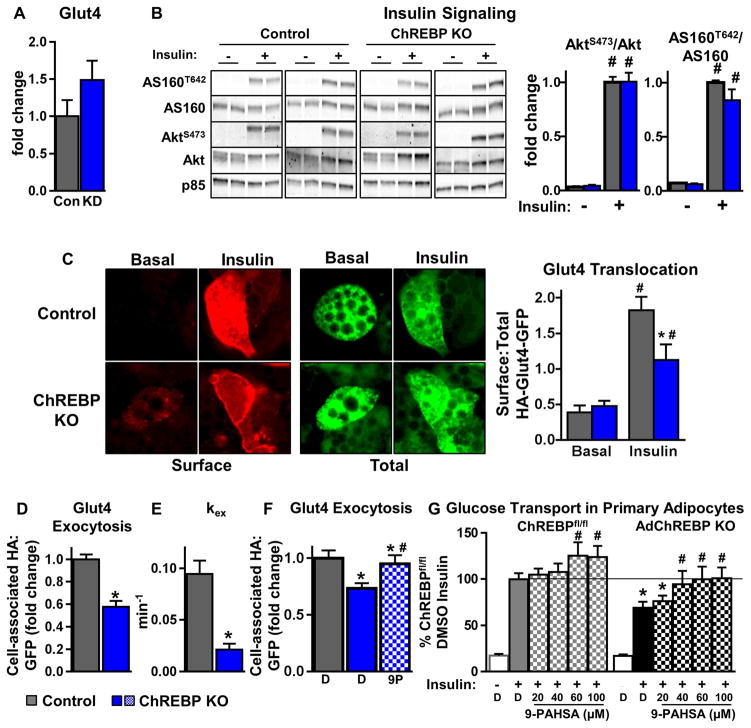
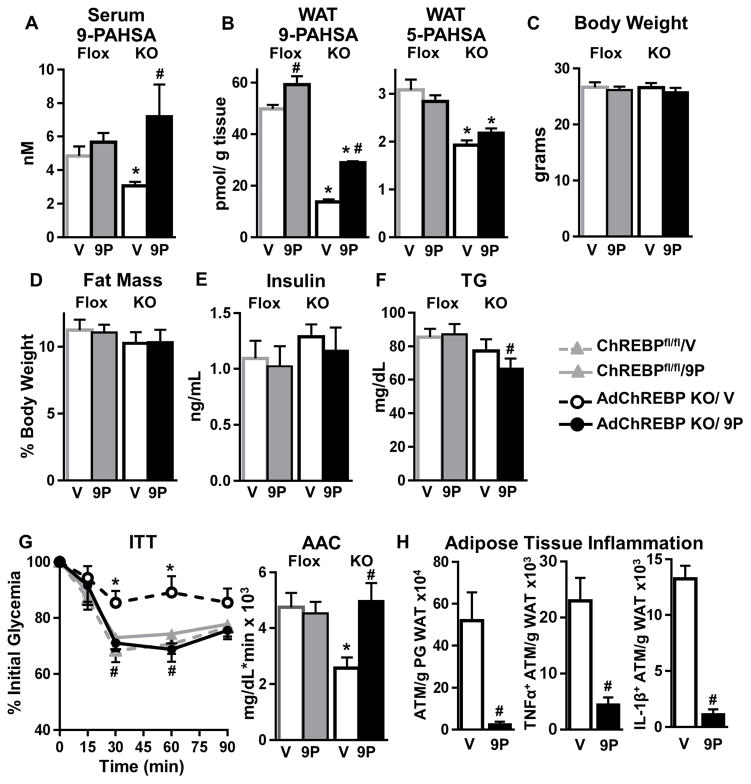
Lower adipose-ChREBP and de novo lipogenesis (DNL) are associated with insulin resistance in humans. Here, we generated adipose-specific ChREBP knockout (AdChREBP KO) mice with negligible sucrose-induced DNL in adipose tissue (AT). Chow-fed AdChREBP KO mice are insulin resistant with impaired insulin action in the liver, muscle, and AT and increased AT inflammation. HFD-fed AdChREBP KO mice are also more insulin resistant than controls. Surprisingly, adipocytes lacking ChREBP display a cell-autonomous reduction in insulin-stimulated glucose transport that is mediated by impaired Glut4 translocation and exocytosis, not lower Glut4 levels. AdChREBP KO mice have lower levels of palmitic acid esters of hydroxy stearic acids (PAHSAs) in serum, and AT. 9-PAHSA supplementation completely rescues their insulin resistance and AT inflammation. 9-PAHSA also normalizes impaired glucose transport and Glut4 exocytosis in ChREBP KO adipocytes. Thus, loss of adipose-ChREBP is sufficient to cause insulin resistance, potentially by regulating AT glucose transport and flux through specific lipogenic pathways.
Keywords: ChREBP; Glut4 trafficking; PAHSA; adipose tissue inflammation; adipose-carbohydrate response element binding protein; de novo lipogenesis; glucose transport; palmitic acid hydroxy stearic acid; systemic insulin resistance.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism.Nature. 2012 Apr 19;484(7394):333-8. doi: 10.1038/nature10986. Nature. 2012. PMID: 22466288 Free PMC article.
-
Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice.Mol Metab. 2017 Nov;6(11):1381-1394. doi: 10.1016/j.molmet.2017.07.006. Epub 2017 Jul 18. Mol Metab. 2017. PMID: 29107286 Free PMC article.
-
Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism.Nat Commun. 2016 Apr 21;7:11365. doi: 10.1038/ncomms11365. Nat Commun. 2016. PMID: 27098609 Free PMC article.
-
GLUT4 Expression in Adipocytes Regulates De Novo Lipogenesis and Levels of a Novel Class of Lipids With Antidiabetic and Anti-inflammatory Effects.Diabetes. 2016 Jul;65(7):1808-15. doi: 10.2337/db16-0221. Epub 2016 Jun 10. Diabetes. 2016. PMID: 27288004 Free PMC article. Review.
-
ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome.Endocr J. 2008 Aug;55(4):617-24. doi: 10.1507/endocrj.k07e-110. Epub 2008 May 19. Endocr J. 2008. PMID: 18490833 Review.
Cited by
-
Lipogenesis inhibitors: therapeutic opportunities and challenges.Nat Rev Drug Discov. 2022 Apr;21(4):283-305. doi: 10.1038/s41573-021-00367-2. Epub 2022 Jan 14. Nat Rev Drug Discov. 2022. PMID: 35031766 Free PMC article. Review.
-
Williams syndrome.Nat Rev Dis Primers. 2021 Jun 17;7(1):42. doi: 10.1038/s41572-021-00276-z. Nat Rev Dis Primers. 2021. PMID: 34140529 Free PMC article. Review.
-
CDK6 inhibits de novo lipogenesis in white adipose tissues but not in the liver.Nat Commun. 2024 Feb 5;15(1):1091. doi: 10.1038/s41467-024-45294-z. Nat Commun. 2024. PMID: 38316780 Free PMC article.
-
Adipose Tissue, Inter-Organ Communication, and the Path to Type 2 Diabetes: The 2016 Banting Medal for Scientific Achievement Lecture.Diabetes. 2019 Jan;68(1):3-14. doi: 10.2337/dbi18-0035. Diabetes. 2019. PMID: 30573674 Free PMC article.
-
Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication.J Lipid Res. 2019 Oct;60(10):1648-1684. doi: 10.1194/jlr.R094060. Epub 2019 Jun 17. J Lipid Res. 2019. PMID: 31209153 Free PMC article. Review.
References
-
- Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. - PubMed
-
- Baraille F, Planchais J, Dentin R, Guilmeau S, Postic C. Integration of ChREBP-Mediated Glucose Sensing into Whole Body Metabolism. Physiology (Bethesda) 2015;30:428–437. - PubMed
-
- Blot V, McGraw TE. Use of quantitative immunofluorescence microscopy to study intracellular trafficking: studies of the GLUT4 glucose transporter. Methods Mol Biol. 2008;457:347–366. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
