

USP7 Is a Tumor-Specific WNT Activator for APC-Mutated Colorectal Cancer by Mediating β-Catenin Deubiquitination
- PMID: 29045831
- PMCID: PMC5656747
- DOI: 10.1016/j.celrep.2017.09.072
USP7 Is a Tumor-Specific WNT Activator for APC-Mutated Colorectal Cancer by Mediating β-Catenin Deubiquitination
Abstract
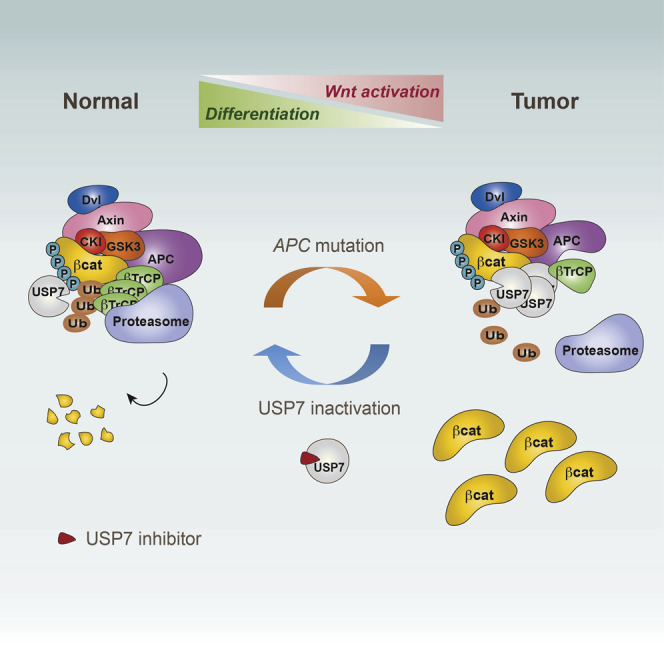
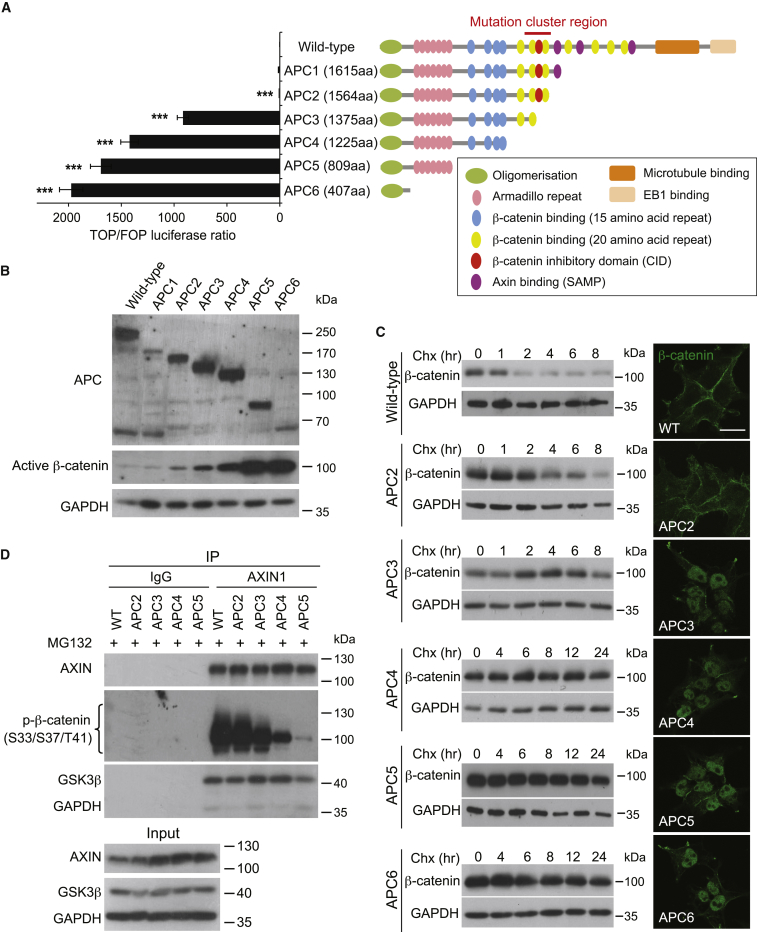
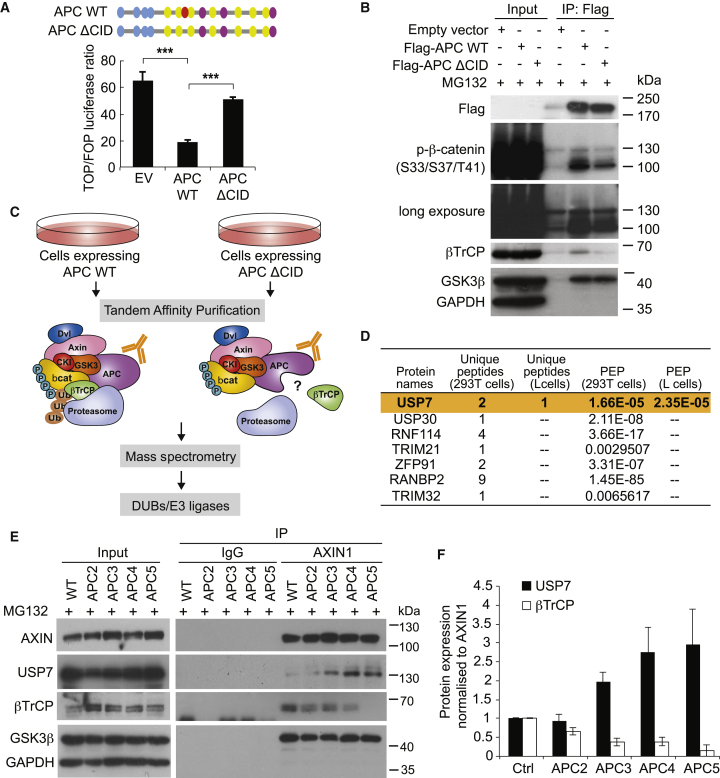
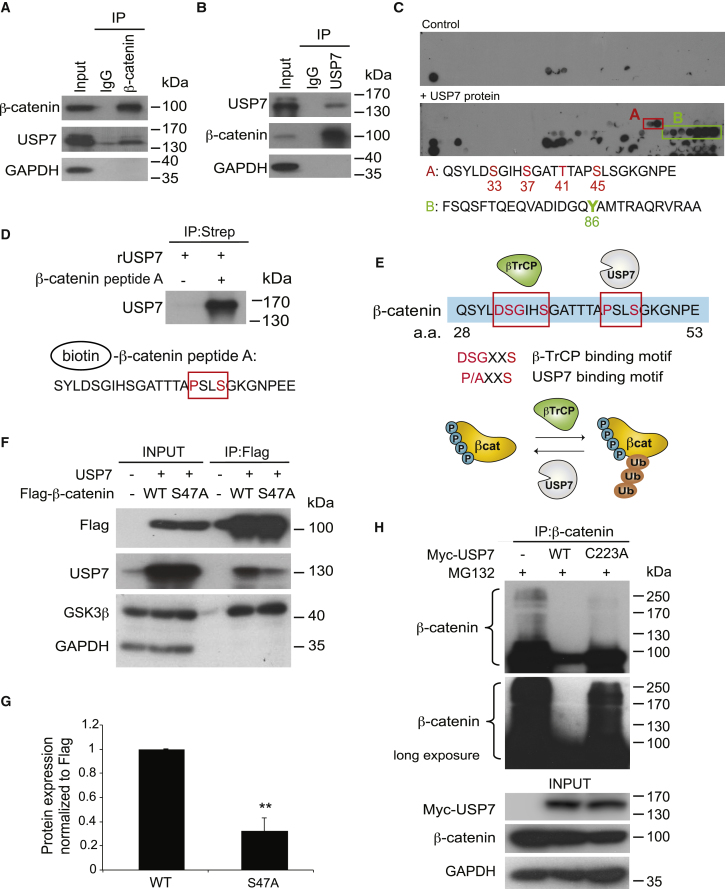
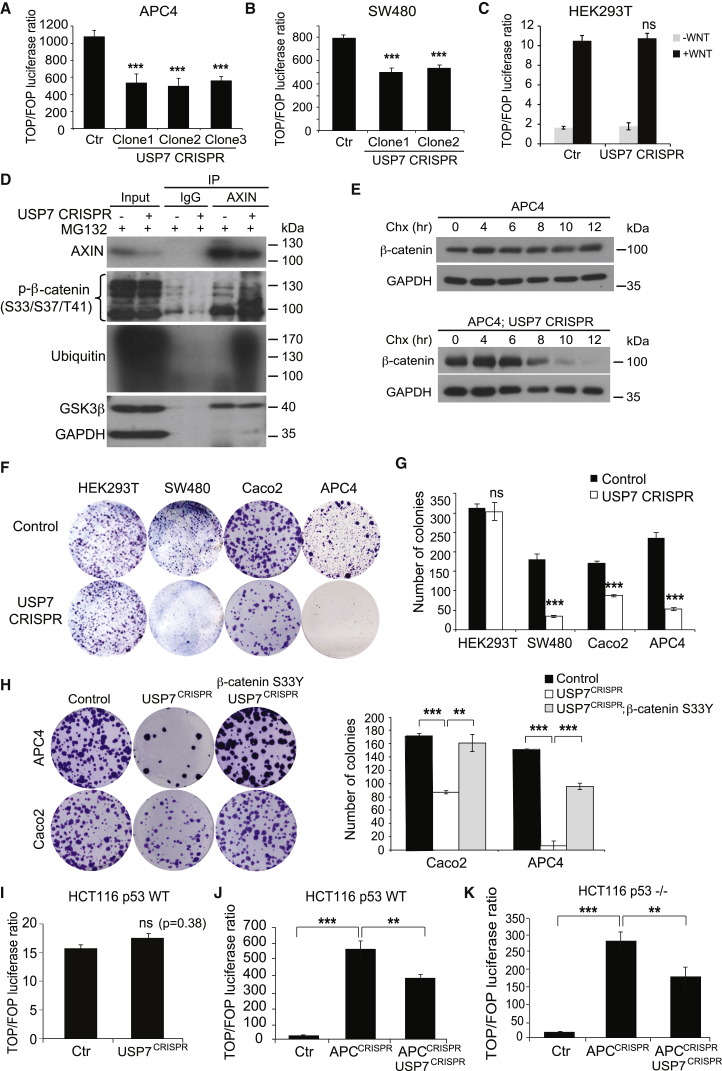
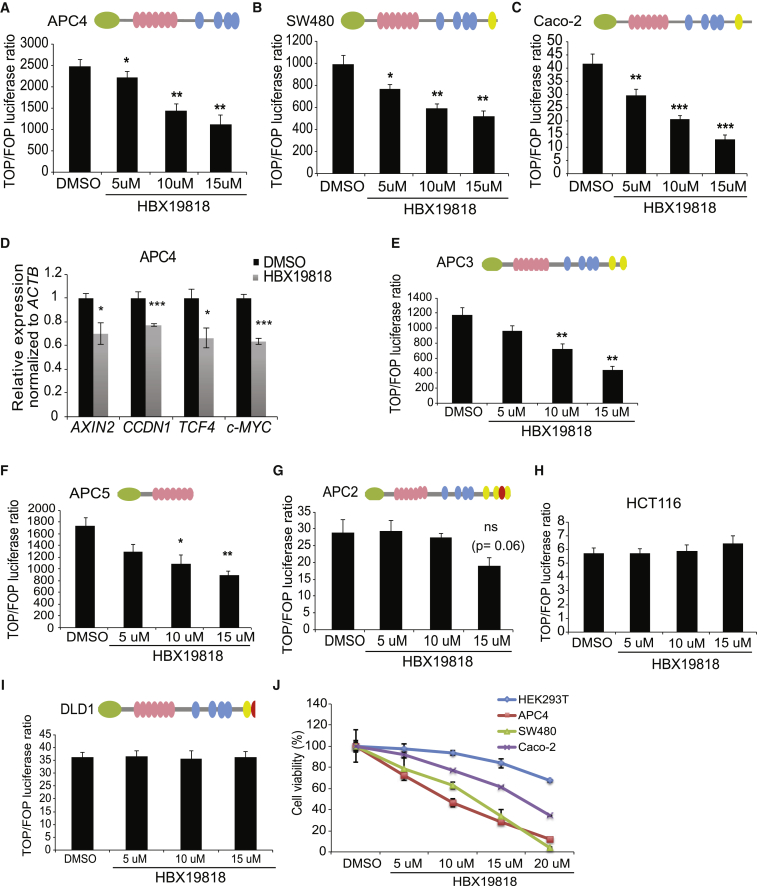
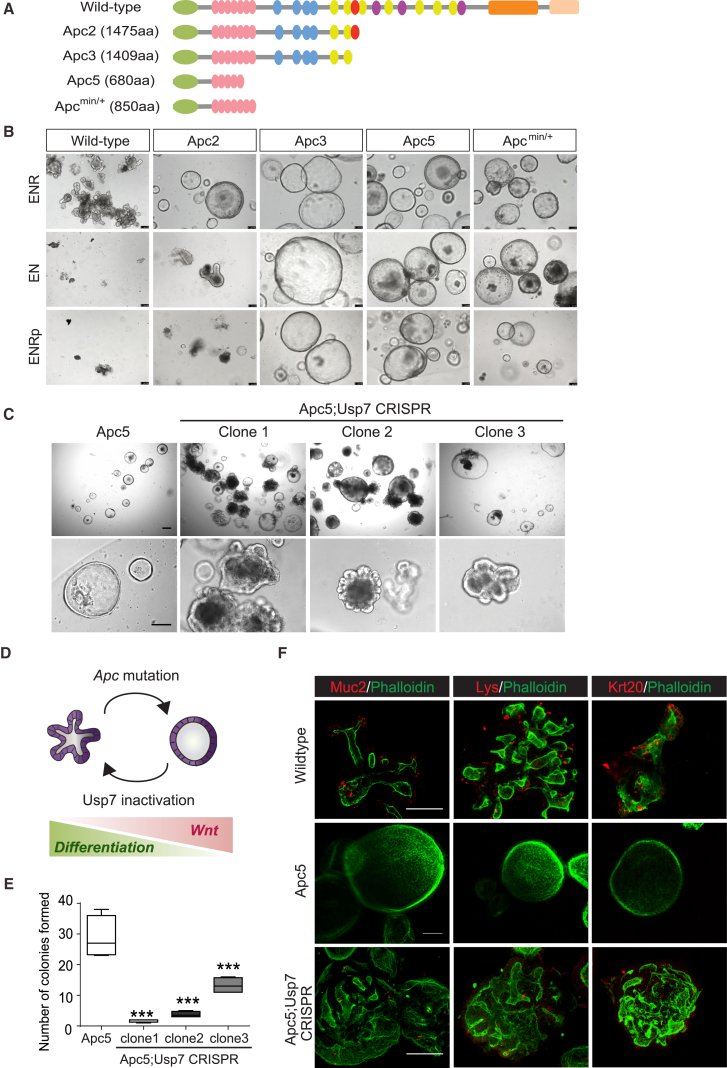
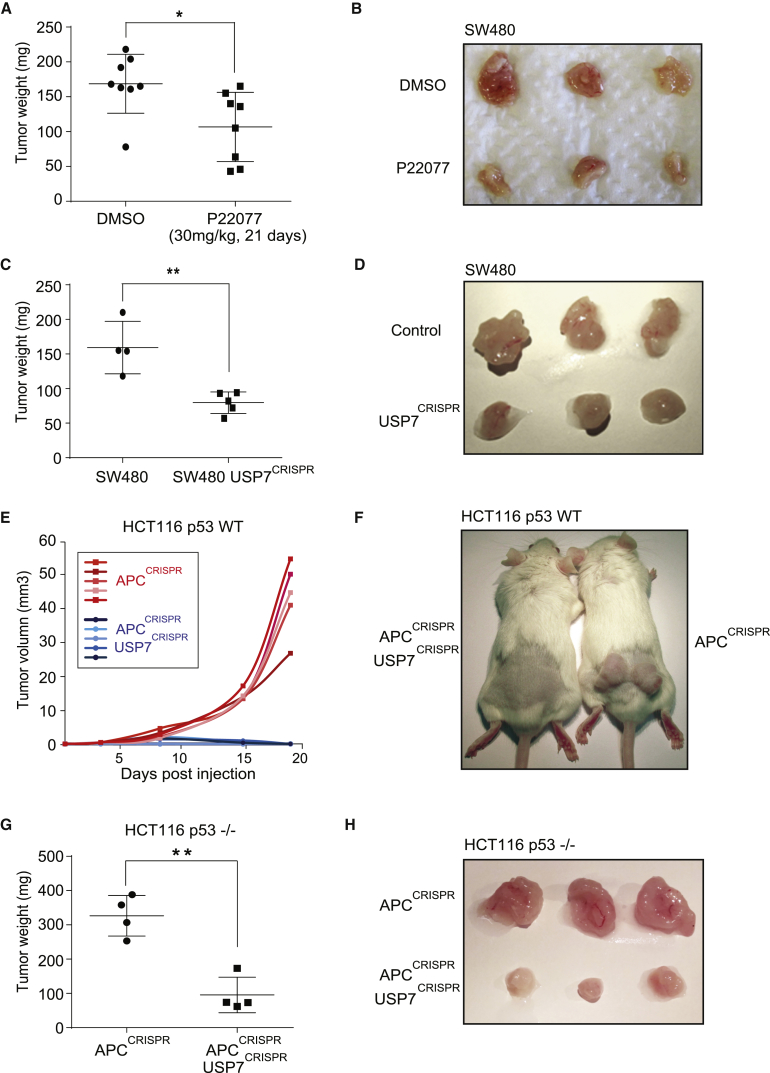
The tumor suppressor gene adenomatous polyposis coli (APC) is mutated in most colorectal cancers (CRCs), resulting in constitutive Wnt activation. To understand the Wnt-activating mechanism of the APC mutation, we applied CRISPR/Cas9 technology to engineer various APC-truncated isogenic lines. We find that the β-catenin inhibitory domain (CID) in APC represents the threshold for pathological levels of Wnt activation and tumor transformation. Mechanistically, CID-deleted APC truncation promotes β-catenin deubiquitination through reverse binding of β-TrCP and USP7 to the destruction complex. USP7 depletion in APC-mutated CRC inhibits Wnt activation by restoring β-catenin ubiquitination, drives differentiation, and suppresses xenograft tumor growth. Finally, the Wnt-activating role of USP7 is specific to APC mutations; thus, it can be used as a tumor-specific therapeutic target for most CRCs.
Keywords: APC; USP7; Wnt signaling; colorectal cancer; ubiquitination; β-catenin.
Copyright © 2017 The Francis Crick Institute. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
USP7 inactivation suppresses APC-mutant intestinal hyperproliferation and tumor development.Stem Cell Reports. 2023 Feb 14;18(2):570-584. doi: 10.1016/j.stemcr.2022.12.013. Epub 2023 Jan 19. Stem Cell Reports. 2023. PMID: 36669491 Free PMC article.
-
Parthenolide inhibits ubiquitin-specific peptidase 7 (USP7), Wnt signaling, and colorectal cancer cell growth.J Biol Chem. 2020 Mar 13;295(11):3576-3589. doi: 10.1074/jbc.RA119.011396. Epub 2020 Feb 6. J Biol Chem. 2020. PMID: 32029476 Free PMC article.
-
USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth.Biochem Pharmacol. 2017 May 1;131:29-39. doi: 10.1016/j.bcp.2017.02.011. Epub 2017 Feb 16. Biochem Pharmacol. 2017. PMID: 28216017
-
Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives.Biochimie. 2019 Feb;157:64-71. doi: 10.1016/j.biochi.2018.11.003. Epub 2018 Nov 8. Biochimie. 2019. PMID: 30414835 Review.
-
Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer.J Natl Cancer Inst. 2017 Aug 1;109(8):djw332. doi: 10.1093/jnci/djw332. J Natl Cancer Inst. 2017. PMID: 28423402 Free PMC article. Review.
Cited by
-
USP10 drives cancer stemness and enables super-competitor signalling in colorectal cancer.Oncogene. 2024 Dec;43(50):3645-3659. doi: 10.1038/s41388-024-03141-x. Epub 2024 Oct 23. Oncogene. 2024. PMID: 39443725 Free PMC article.
-
Deubiquitylating Enzymes in Cancer and Immunity.Adv Sci (Weinh). 2023 Dec;10(36):e2303807. doi: 10.1002/advs.202303807. Epub 2023 Oct 27. Adv Sci (Weinh). 2023. PMID: 37888853 Free PMC article. Review.
-
Circular RNAs in hepatocellular carcinoma: biogenesis, function, and pathology.Front Genet. 2023 Jul 7;14:1106665. doi: 10.3389/fgene.2023.1106665. eCollection 2023. Front Genet. 2023. PMID: 37485335 Free PMC article. Review.
-
Ubiquitin-Specific Proteases: Players in Cancer Cellular Processes.Pharmaceuticals (Basel). 2021 Aug 26;14(9):848. doi: 10.3390/ph14090848. Pharmaceuticals (Basel). 2021. PMID: 34577547 Free PMC article. Review.
-
Cachd1 interacts with Wnt receptors and regulates neuronal asymmetry in the zebrafish brain.Science. 2024 May 3;384(6695):573-579. doi: 10.1126/science.ade6970. Epub 2024 May 2. Science. 2024. PMID: 38696577 Free PMC article.
References
-
- Clevers H., Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
