

Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition
- PMID: 28882883
- PMCID: PMC5731086
- DOI: 10.1182/blood-2017-05-782102
Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition
Abstract
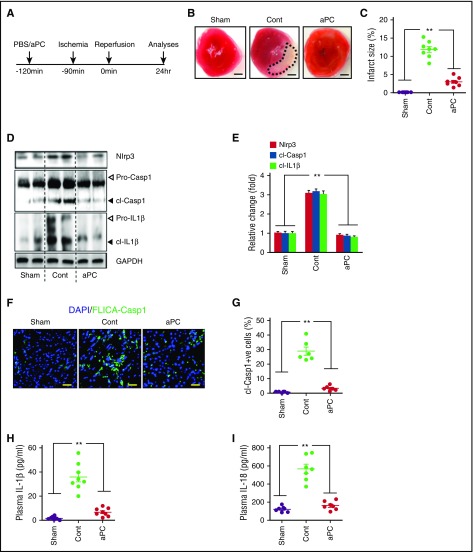
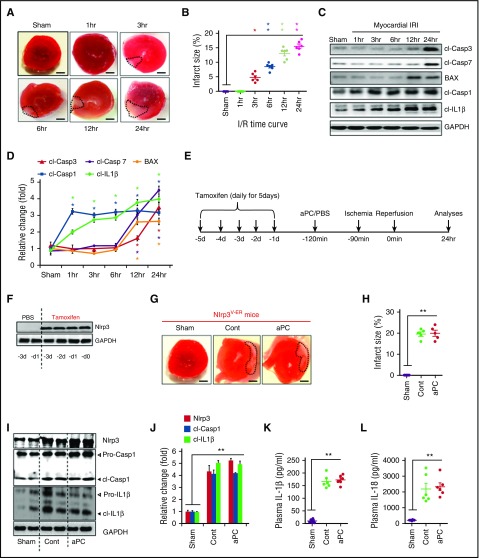
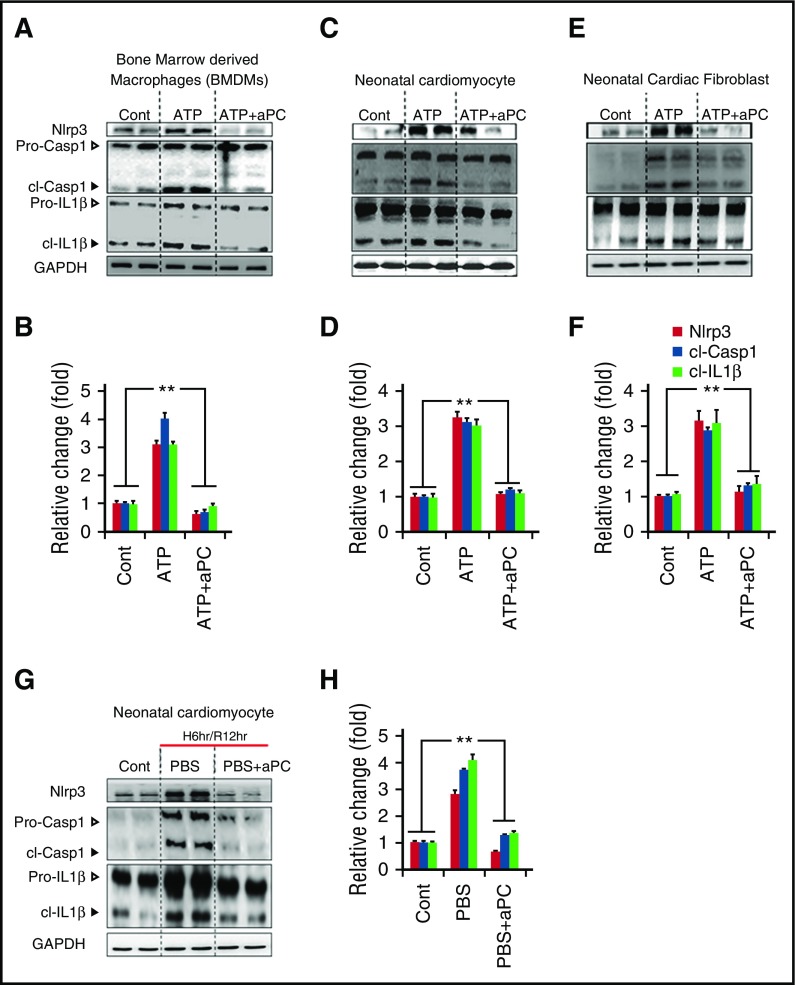
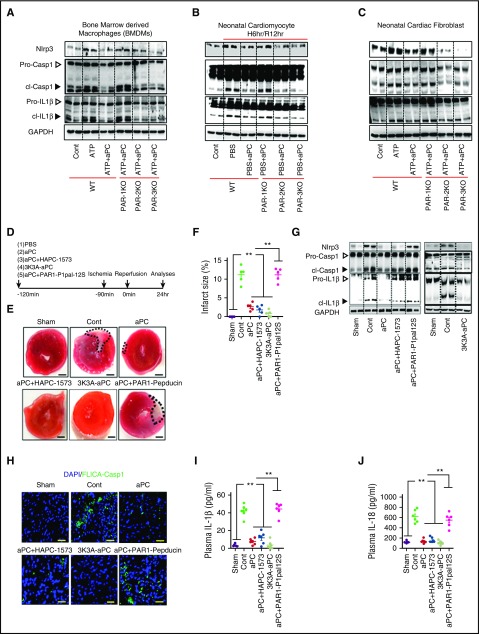
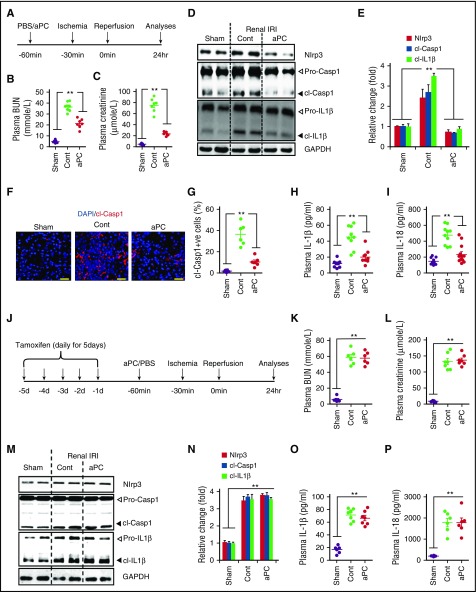
Cytoprotection by activated protein C (aPC) after ischemia-reperfusion injury (IRI) is associated with apoptosis inhibition. However, IRI is hallmarked by inflammation, and hence, cell-death forms disjunct from immunologically silent apoptosis are, in theory, more likely to be relevant. Because pyroptosis (ie, cell death resulting from inflammasome activation) is typically observed in IRI, we speculated that aPC ameliorates IRI by inhibiting inflammasome activation. Here we analyzed the impact of aPC on inflammasome activity in myocardial and renal IRIs. aPC treatment before or after myocardial IRI reduced infarct size and Nlrp3 inflammasome activation in mice. Kinetic in vivo analyses revealed that Nlrp3 inflammasome activation preceded myocardial injury and apoptosis, corroborating a pathogenic role of the Nlrp3 inflammasome. The constitutively active Nlrp3A350V mutation abolished the protective effect of aPC, demonstrating that Nlrp3 suppression is required for aPC-mediated protection from IRI. In vitro aPC inhibited inflammasome activation in macrophages, cardiomyocytes, and cardiac fibroblasts via proteinase-activated receptor 1 (PAR-1) and mammalian target of rapamycin complex 1 (mTORC1) signaling. Accordingly, inhibiting PAR-1 signaling, but not the anticoagulant properties of aPC, abolished the ability of aPC to restrict Nlrp3 inflammasome activity and tissue damage in myocardial IRI. Targeting biased PAR-1 signaling via parmodulin-2 restricted mTORC1 and Nlrp3 inflammasome activation and limited myocardial IRI as efficiently as aPC. The relevance of aPC-mediated Nlrp3 inflammasome suppression after IRI was corroborated in renal IRI, where the tissue protective effect of aPC was likewise dependent on Nlrp3 inflammasome suppression. These studies reveal that aPC protects from IRI by restricting mTORC1-dependent inflammasome activation and that mimicking biased aPC PAR-1 signaling using parmodulins may be a feasible therapeutic approach to combat IRI.
© 2017 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: C.D. is an inventor on a patent (WO2012/040636) describing parmodulin-2 (ML161). The remaining authors declare no competing financial interests.
Figures


Comment in
-
Inflammation: Activated protein C inhibits inflammasome activation in IRI.Nat Rev Nephrol. 2017 Nov;13(11):662. doi: 10.1038/nrneph.2017.139. Epub 2017 Sep 25. Nat Rev Nephrol. 2017. PMID: 28944779 No abstract available.
-
On PAR with aPC to target inflammasomes.Blood. 2017 Dec 14;130(24):2579-2581. doi: 10.1182/blood-2017-09-806307. Blood. 2017. PMID: 29242206 No abstract available.
Similar articles
-
Scutellarin protects against myocardial ischemia-reperfusion injury by suppressing NLRP3 inflammasome activation.Phytomedicine. 2020 Mar;68:153169. doi: 10.1016/j.phymed.2020.153169. Epub 2020 Jan 16. Phytomedicine. 2020. PMID: 31999976
-
Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway.Aging (Albany NY). 2020 Nov 24;12(23):24270-24287. doi: 10.18632/aging.202143. Epub 2020 Nov 24. Aging (Albany NY). 2020. PMID: 33232283 Free PMC article.
-
Naoxintong attenuates Ischaemia/reperfusion Injury through inhibiting NLRP3 inflammasome activation.J Cell Mol Med. 2017 Jan;21(1):4-12. doi: 10.1111/jcmm.12915. Epub 2016 Oct 26. J Cell Mol Med. 2017. PMID: 27785882 Free PMC article.
-
NLRP3 inflammasome: A potential therapeutic target to minimize renal ischemia/reperfusion injury during transplantation.Transpl Immunol. 2022 Dec;75:101718. doi: 10.1016/j.trim.2022.101718. Epub 2022 Sep 17. Transpl Immunol. 2022. PMID: 36126906 Review.
-
Cell-Specific Roles of NLRP3 Inflammasome in Myocardial Infarction.J Cardiovasc Pharmacol. 2019 Sep;74(3):188-193. doi: 10.1097/FJC.0000000000000709. J Cardiovasc Pharmacol. 2019. PMID: 31356542 Review.
Cited by
-
Activated protein C in neuroprotection and malaria.Curr Opin Hematol. 2019 Sep;26(5):320-330. doi: 10.1097/MOH.0000000000000528. Curr Opin Hematol. 2019. PMID: 31348046 Free PMC article. Review.
-
Knockdown of ELF4 aggravates renal injury in ischemia/reperfusion mice through promotion of pyroptosis, inflammation, oxidative stress, and endoplasmic reticulum stress.BMC Mol Cell Biol. 2023 Jul 20;24(1):22. doi: 10.1186/s12860-023-00485-2. BMC Mol Cell Biol. 2023. PMID: 37474923 Free PMC article.
-
mTORC1 beyond anabolic metabolism: Regulation of cell death.J Cell Biol. 2022 Dec 5;221(12):e202208103. doi: 10.1083/jcb.202208103. Epub 2022 Oct 25. J Cell Biol. 2022. PMID: 36282248 Free PMC article. Review.
-
NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions.Front Immunol. 2021 May 28;12:683803. doi: 10.3389/fimmu.2021.683803. eCollection 2021. Front Immunol. 2021. PMID: 34122445 Free PMC article.
-
Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis.Clin Transl Med. 2023 Jan;13(1):e1170. doi: 10.1002/ctm2.1170. Clin Transl Med. 2023. PMID: 36629024 Free PMC article. Review.
References
-
- Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8(5):292-300. - PubMed
-
- Timmers L, Pasterkamp G, de Hoog VC, Arslan F, Appelman Y, de Kleijn DP. The innate immune response in reperfused myocardium. Cardiovasc Res. 2012;94(2):276-283. - PubMed
-
- Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM. Inflammation in myocardial diseases. Circ Res. 2012;110(1):126-144. - PubMed
-
- Abbate A, Salloum FN, Vecile E, et al. . Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117(20):2670-2683. - PubMed
-
- Salloum FN, Chau V, Varma A, et al. . Anakinra in experimental acute myocardial infarction--does dosage or duration of treatment matter? Cardiovasc Drugs Ther. 2009;23(2):129-135. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
