

Regulation of rice root development by a retrotransposon acting as a microRNA sponge
- PMID: 28847366
- PMCID: PMC5599236
- DOI: 10.7554/eLife.30038
Regulation of rice root development by a retrotransposon acting as a microRNA sponge
Abstract
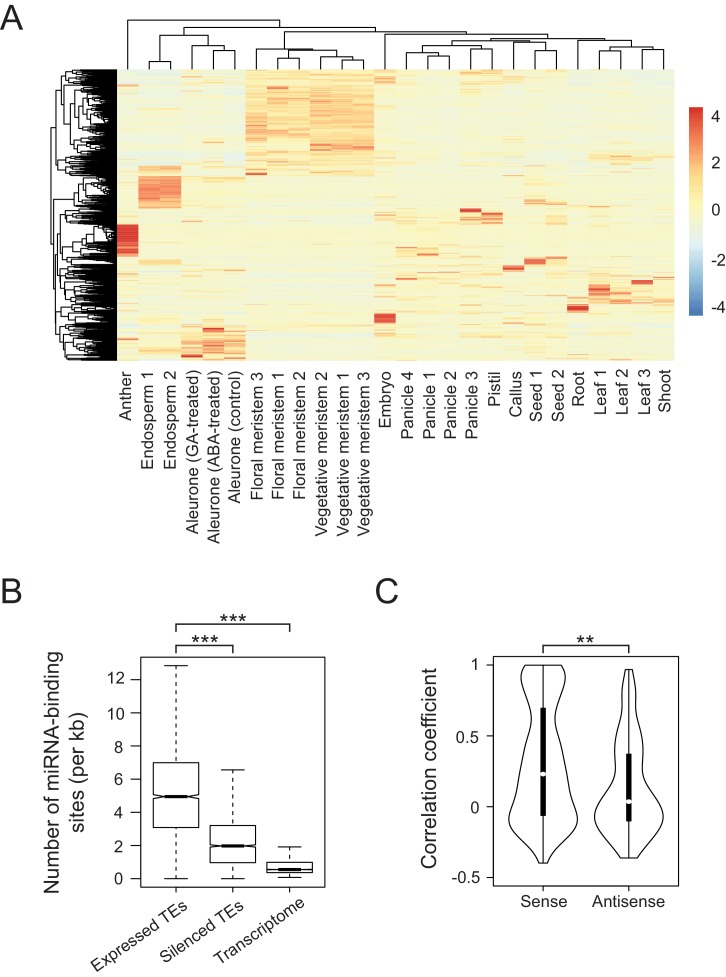
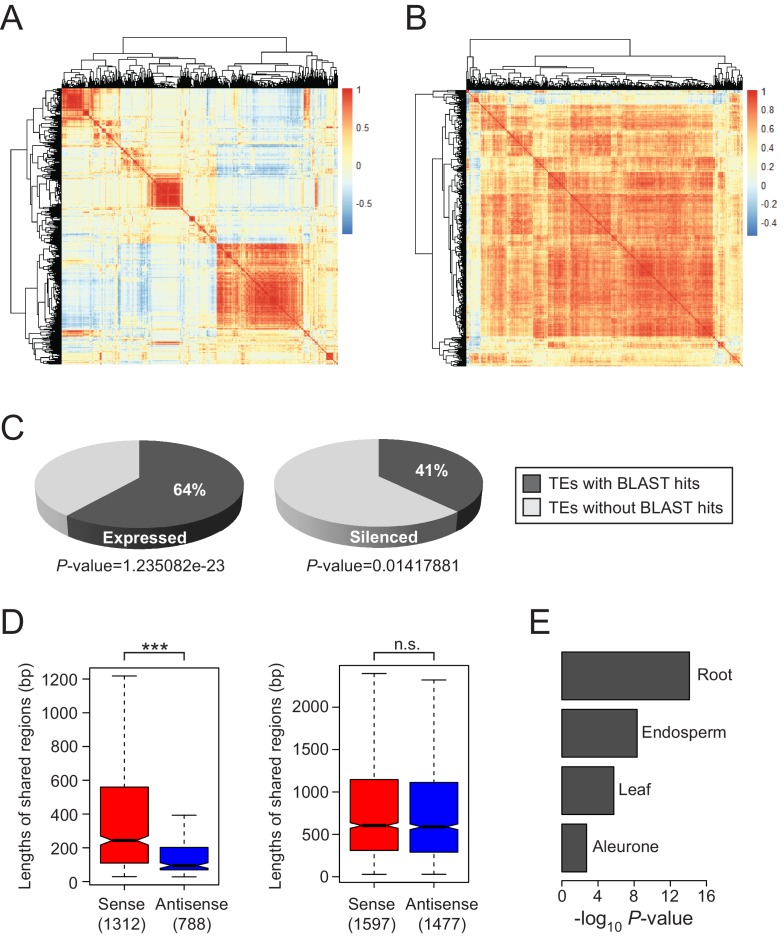
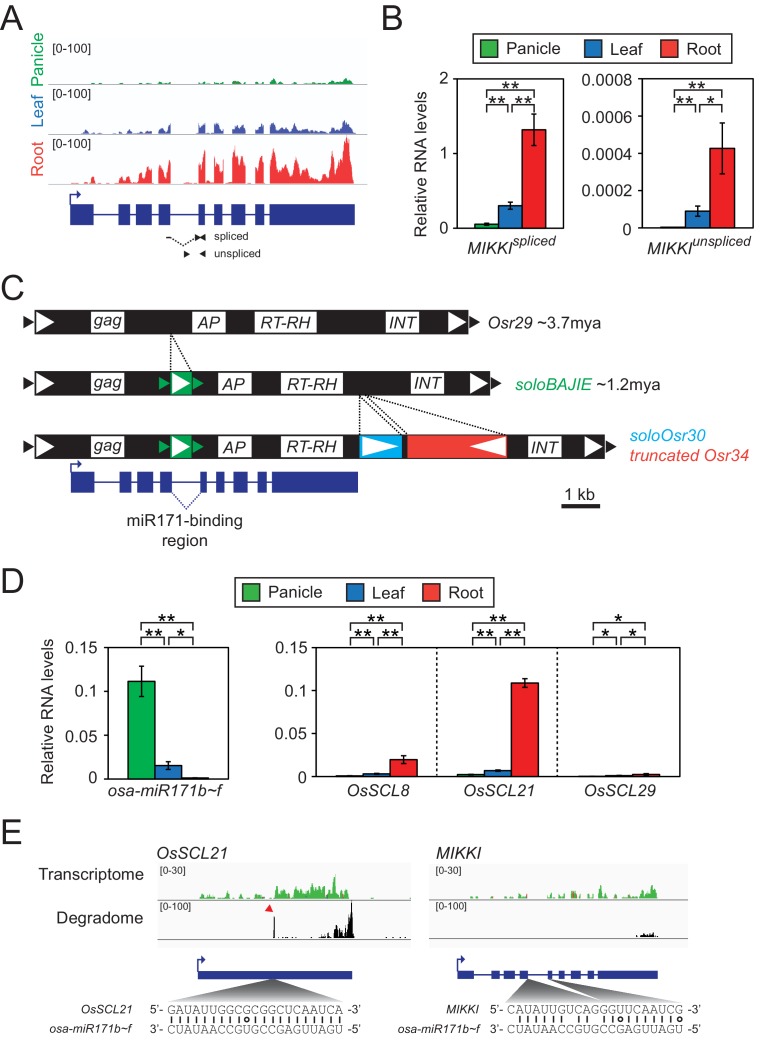
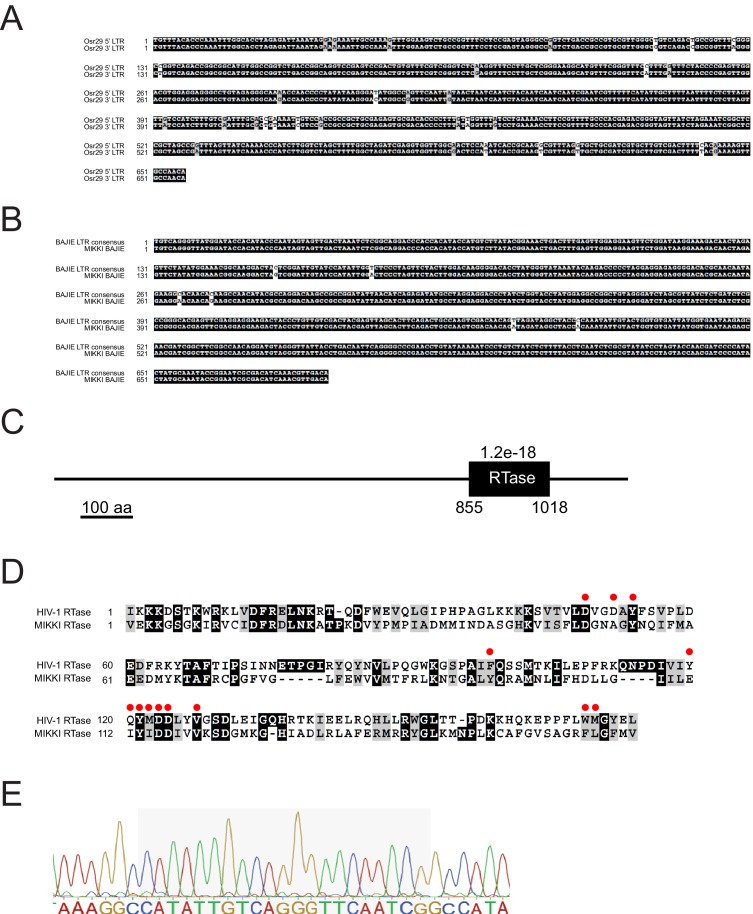
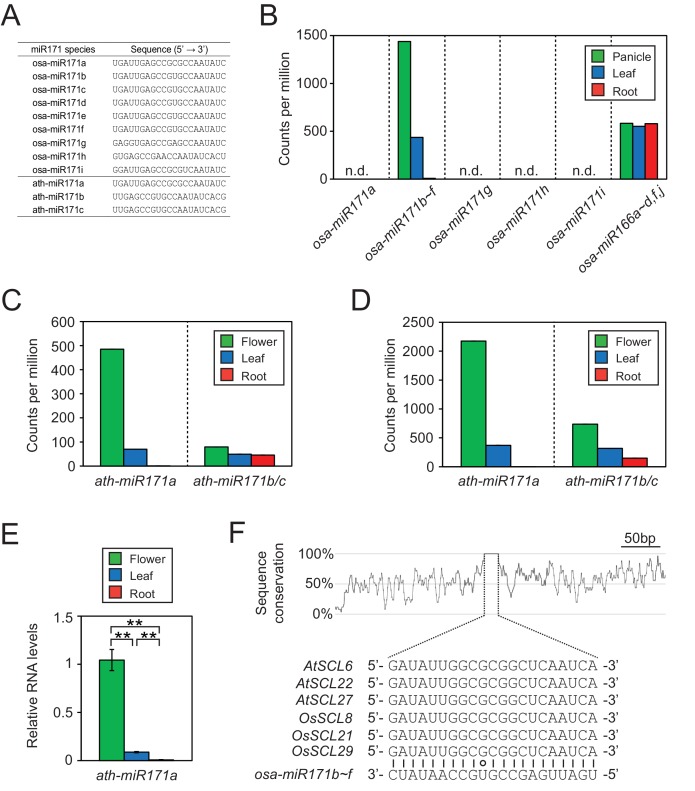
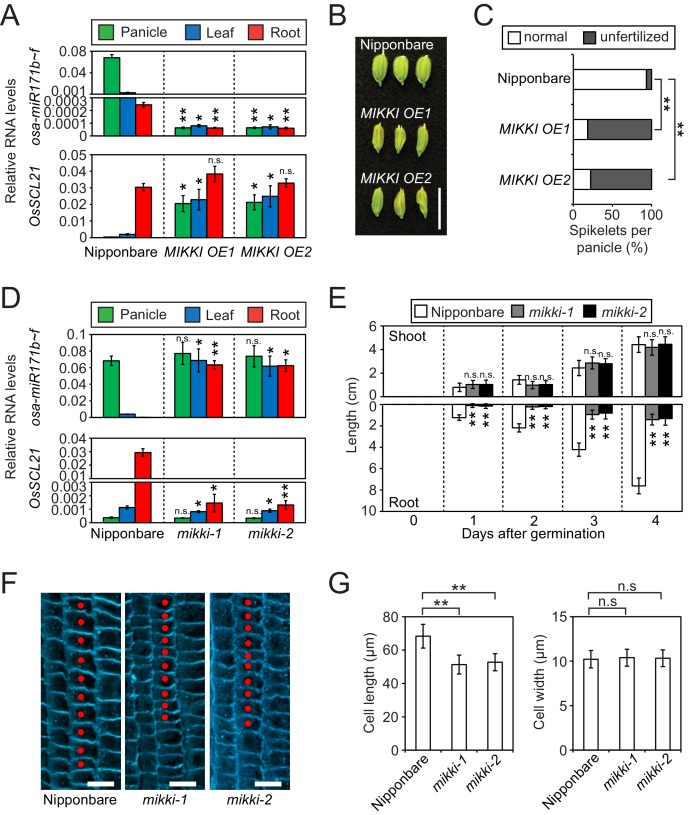
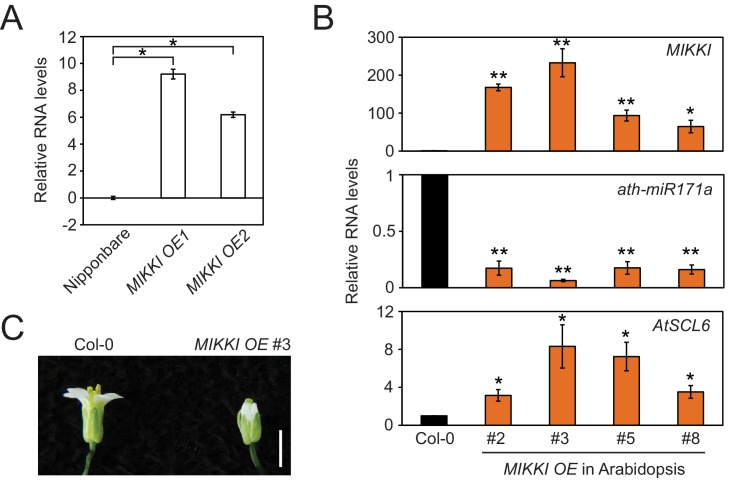
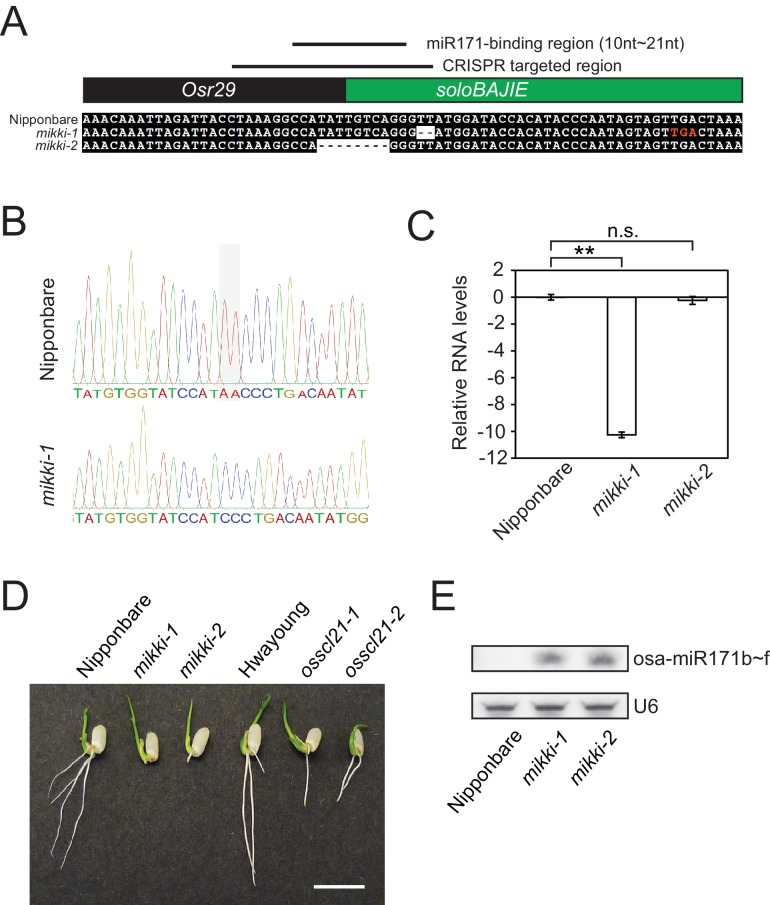
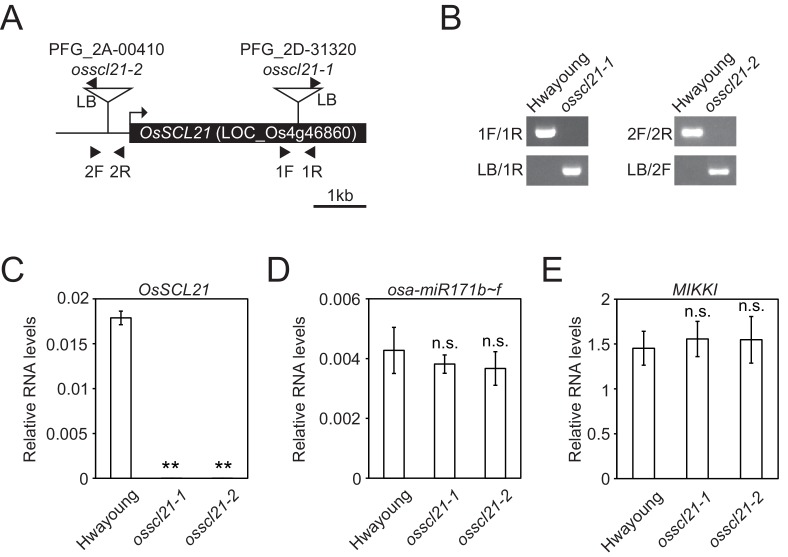
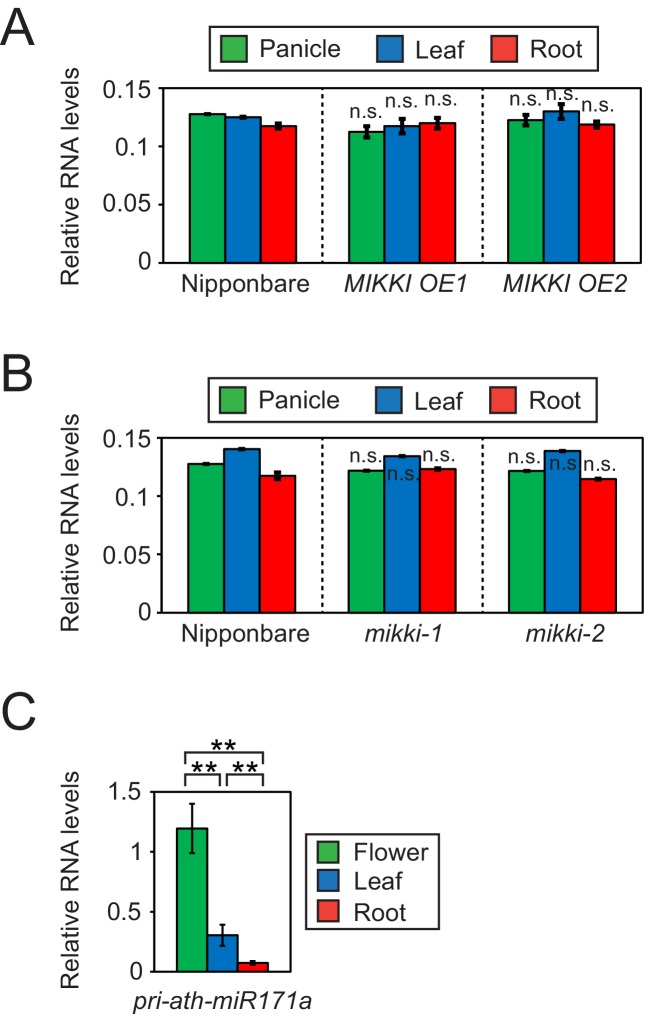
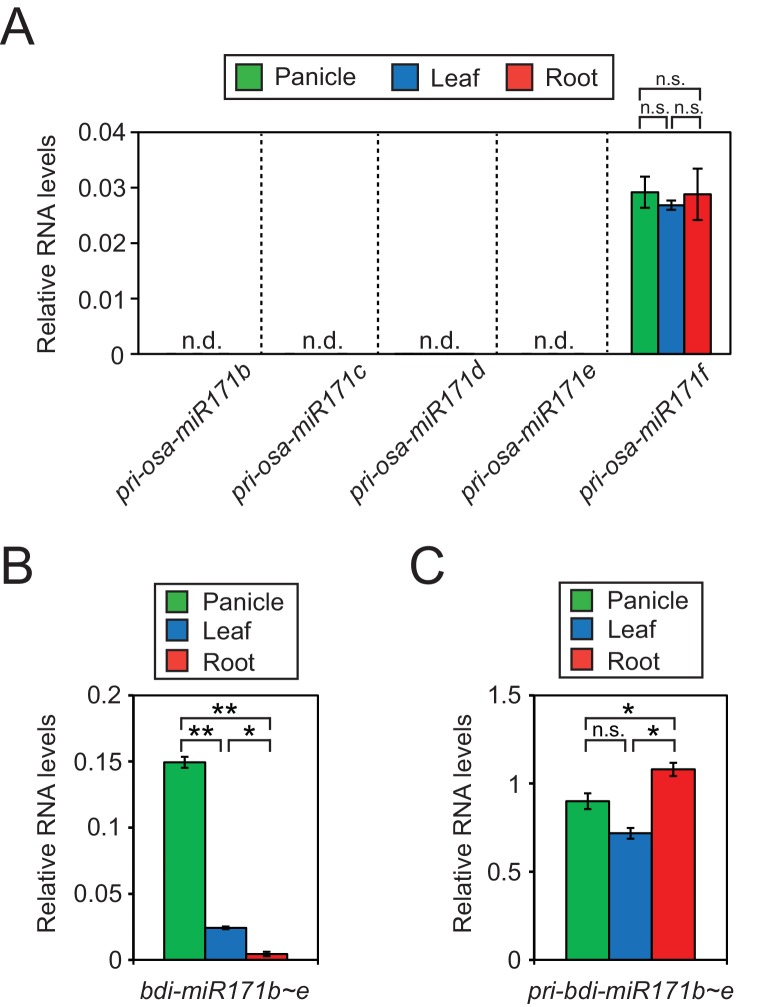
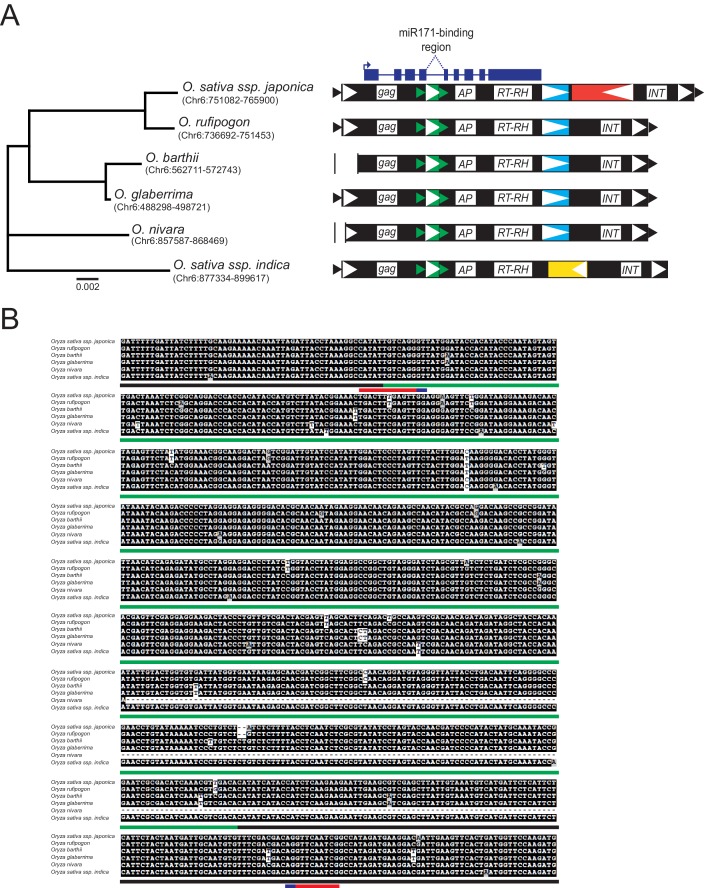
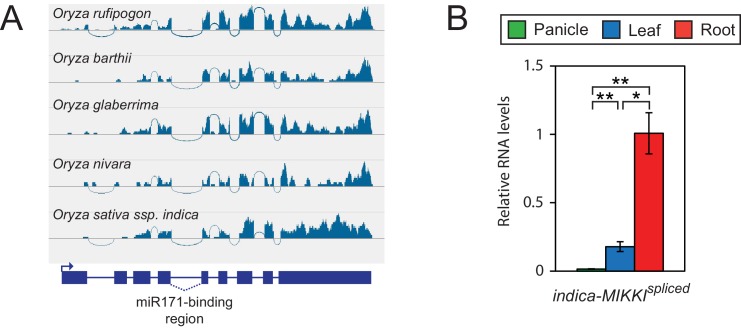
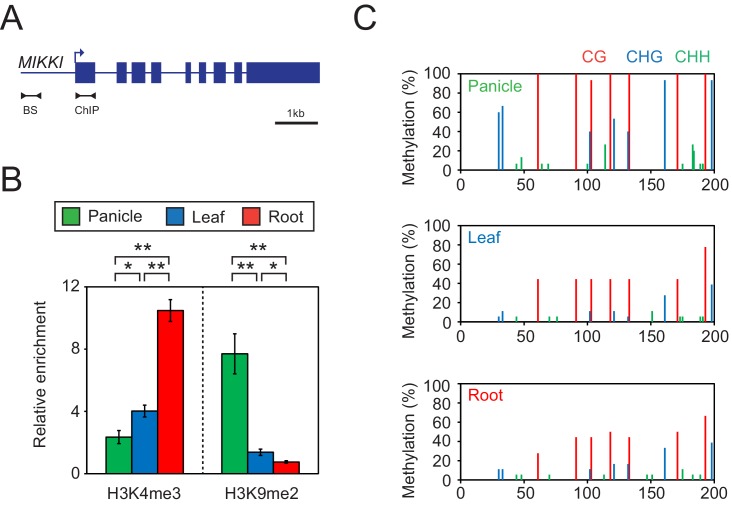
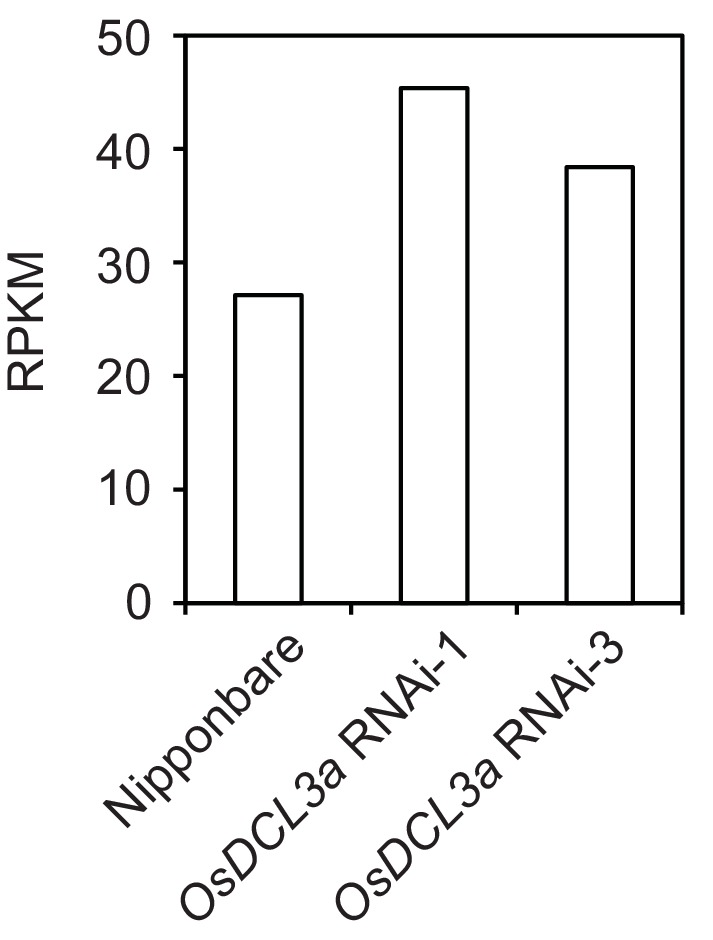
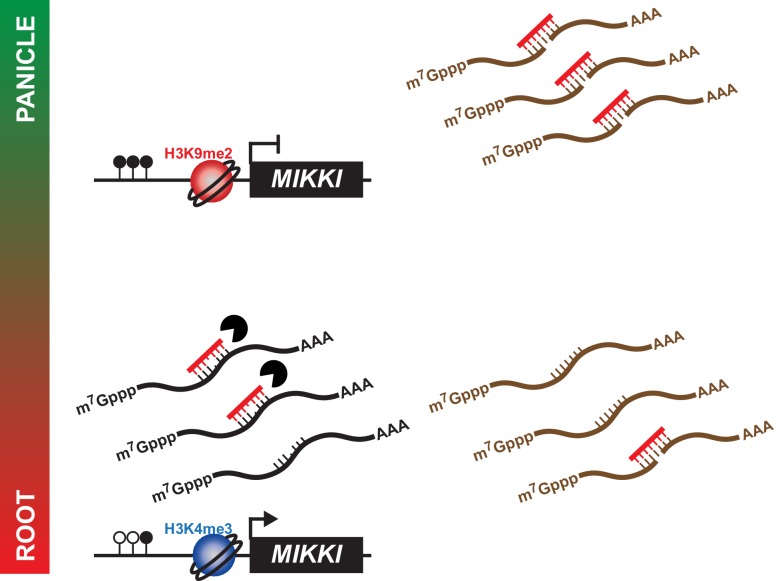
It is well documented that transposable elements (TEs) can regulate the expression of neighbouring genes. However, their ability to act in trans and influence ectopic loci has been reported rarely. We searched in rice transcriptomes for tissue-specific expression of TEs and found them to be regulated developmentally. They often shared sequence homology with co-expressed genes and contained potential microRNA-binding sites, which suggested possible contributions to gene regulation. In fact, we have identified a retrotransposon that is highly transcribed in roots and whose spliced transcript constitutes a target mimic for miR171. miR171 destabilizes mRNAs encoding the root-specific family of SCARECROW-Like transcription factors. We demonstrate that retrotransposon-derived transcripts act as decoys for miR171, triggering its degradation and thus results in the root-specific accumulation of SCARECROW-Like mRNAs. Such transposon-mediated post-transcriptional control of miR171 levels is conserved in diverse rice species.
Keywords: Oryza sativa; miR171; plant biology; root development.
Conflict of interest statement
No competing interests declared.
Figures
Similar articles
-
MicroRNA Sequencing Revealed Citrus Adaptation to Long-Term Boron Toxicity through Modulation of Root Development by miR319 and miR171.Int J Mol Sci. 2019 Mar 21;20(6):1422. doi: 10.3390/ijms20061422. Int J Mol Sci. 2019. PMID: 30901819 Free PMC article.
-
Distinctive expression patterns and roles of the miRNA393/TIR1 homolog module in regulating flag leaf inclination and primary and crown root growth in rice (Oryza sativa).New Phytol. 2012 Oct;196(1):149-161. doi: 10.1111/j.1469-8137.2012.04248.x. Epub 2012 Jul 27. New Phytol. 2012. PMID: 22846038
-
Identification of CROWN ROOTLESS1-regulated genes in rice reveals specific and conserved elements of postembryonic root formation.New Phytol. 2015 Apr;206(1):243-254. doi: 10.1111/nph.13196. Epub 2014 Dec 1. New Phytol. 2015. PMID: 25442012
-
Transposon-Derived Non-coding RNAs and Their Function in Plants.Front Plant Sci. 2018 May 3;9:600. doi: 10.3389/fpls.2018.00600. eCollection 2018. Front Plant Sci. 2018. PMID: 29774045 Free PMC article. Review.
-
Role of microRNA miR171 in plant development.PeerJ. 2023 Jul 10;11:e15632. doi: 10.7717/peerj.15632. eCollection 2023. PeerJ. 2023. PMID: 37456878 Free PMC article. Review.
Cited by
-
The plant noncoding transcriptome: a versatile environmental sensor.EMBO J. 2023 Oct 16;42(20):e114400. doi: 10.15252/embj.2023114400. Epub 2023 Sep 21. EMBO J. 2023. PMID: 37735935 Free PMC article. Review.
-
Small But Powerful: MicroRNA-Derived Peptides Promote Grape Adventitious Root Formation.Plant Physiol. 2020 Jun;183(2):429-430. doi: 10.1104/pp.20.00515. Plant Physiol. 2020. PMID: 32493803 Free PMC article. No abstract available.
-
Regulatory non-coding RNAs: a new frontier in regulation of plant biology.Funct Integr Genomics. 2021 Jul;21(3-4):313-330. doi: 10.1007/s10142-021-00787-8. Epub 2021 May 20. Funct Integr Genomics. 2021. PMID: 34013486 Free PMC article. Review.
-
Long non-coding RNAs: Fine-tuning the developmental responses in plants.J Biosci. 2019 Sep;44(4):77. J Biosci. 2019. PMID: 31502555 Review.
-
The landscape of transposable elements and satellite DNAs in the genome of a dioecious plant spinach (Spinacia oleracea L.).Mob DNA. 2019 Jan 18;10:3. doi: 10.1186/s13100-019-0147-6. eCollection 2019. Mob DNA. 2019. PMID: 30675191 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
