

Regulation of type I interferon responses by mitochondria-derived reactive oxygen species in plasmacytoid dendritic cells
- PMID: 28818792
- PMCID: PMC5558471
- DOI: 10.1016/j.redox.2017.07.016
Regulation of type I interferon responses by mitochondria-derived reactive oxygen species in plasmacytoid dendritic cells
Abstract
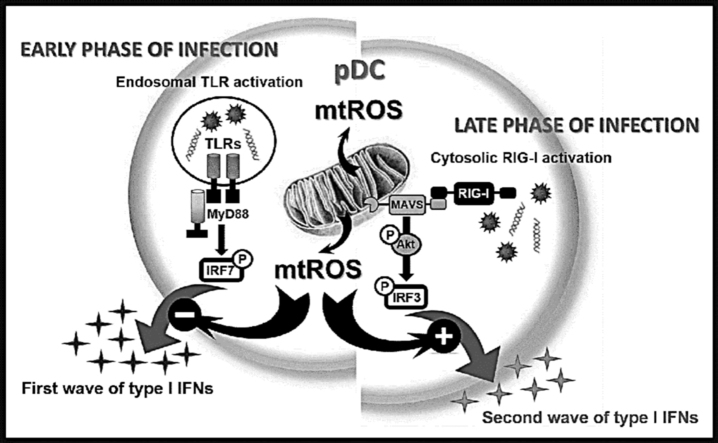
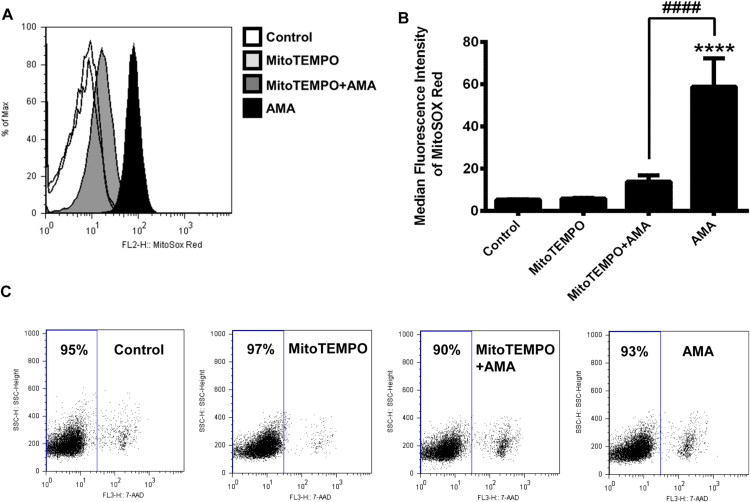
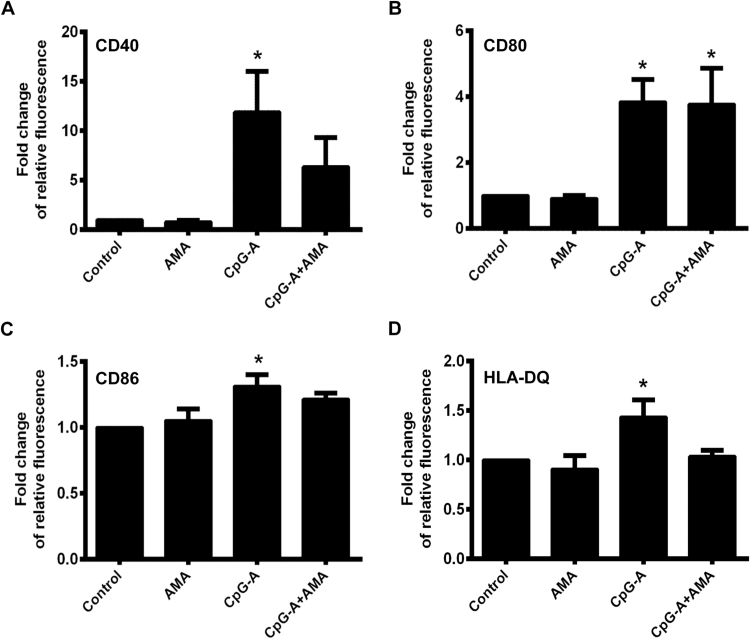
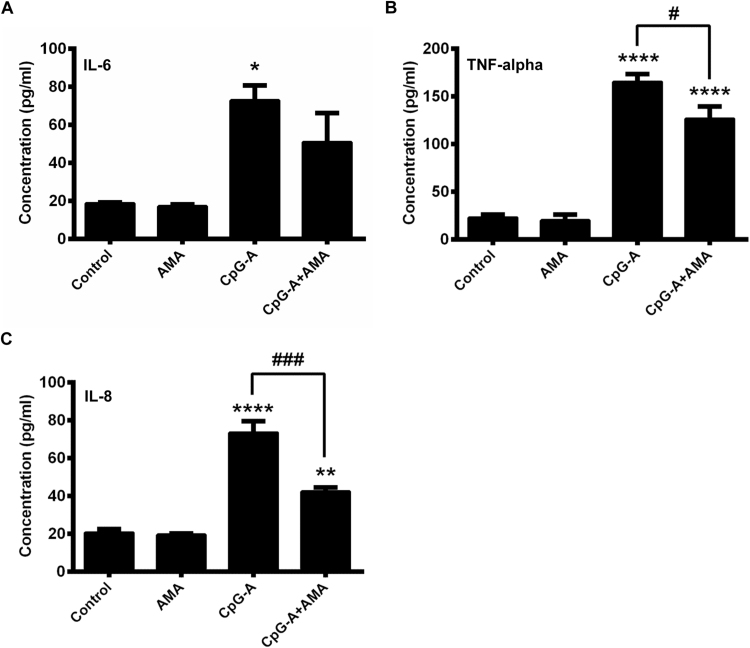
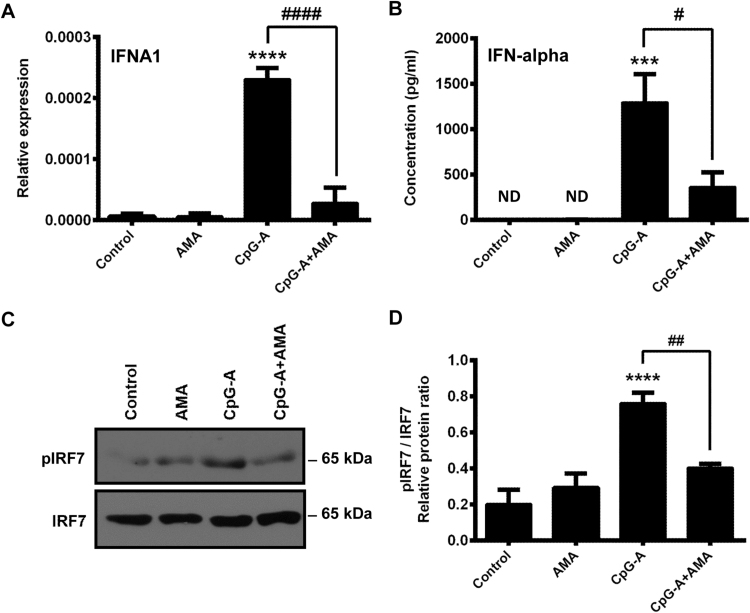
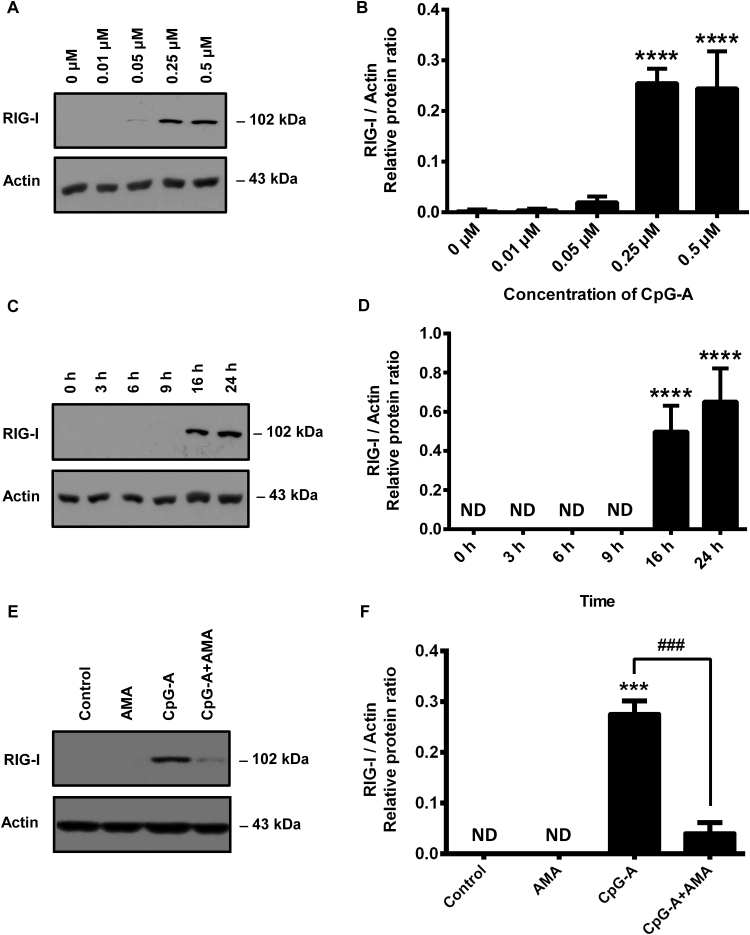
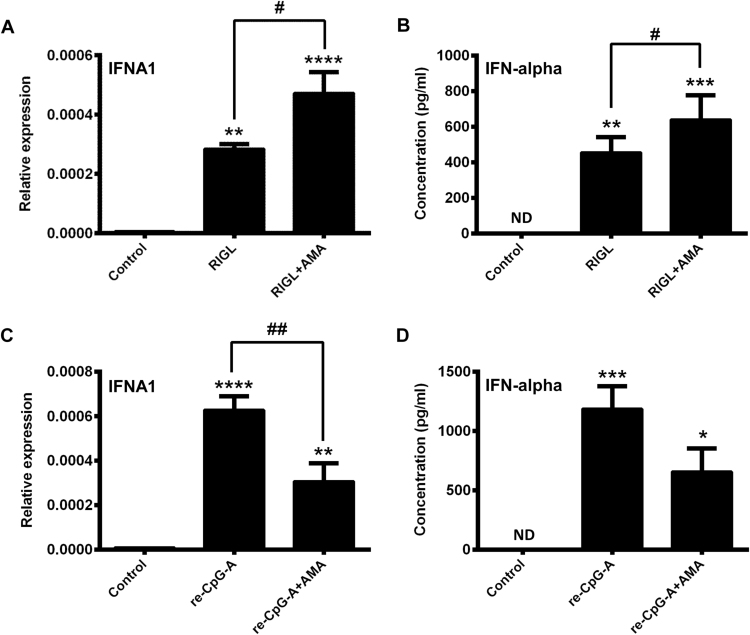
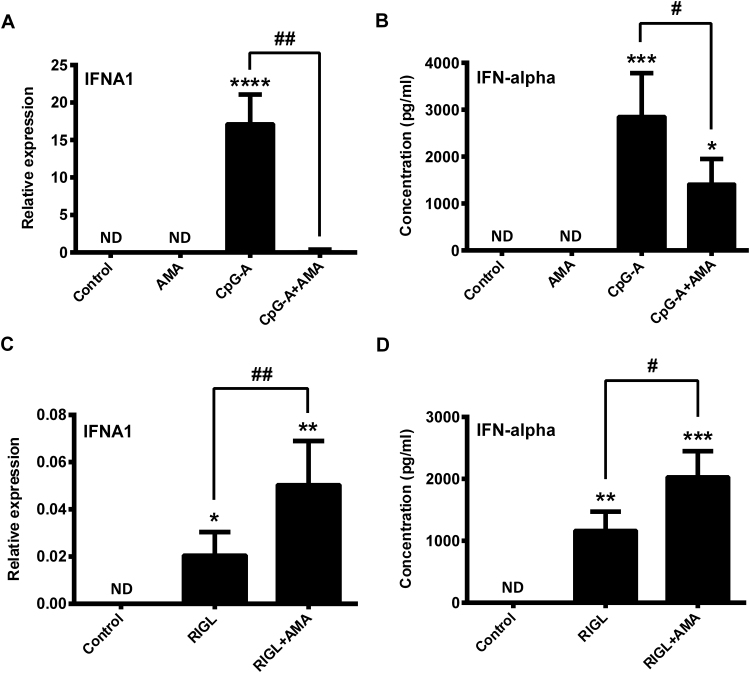
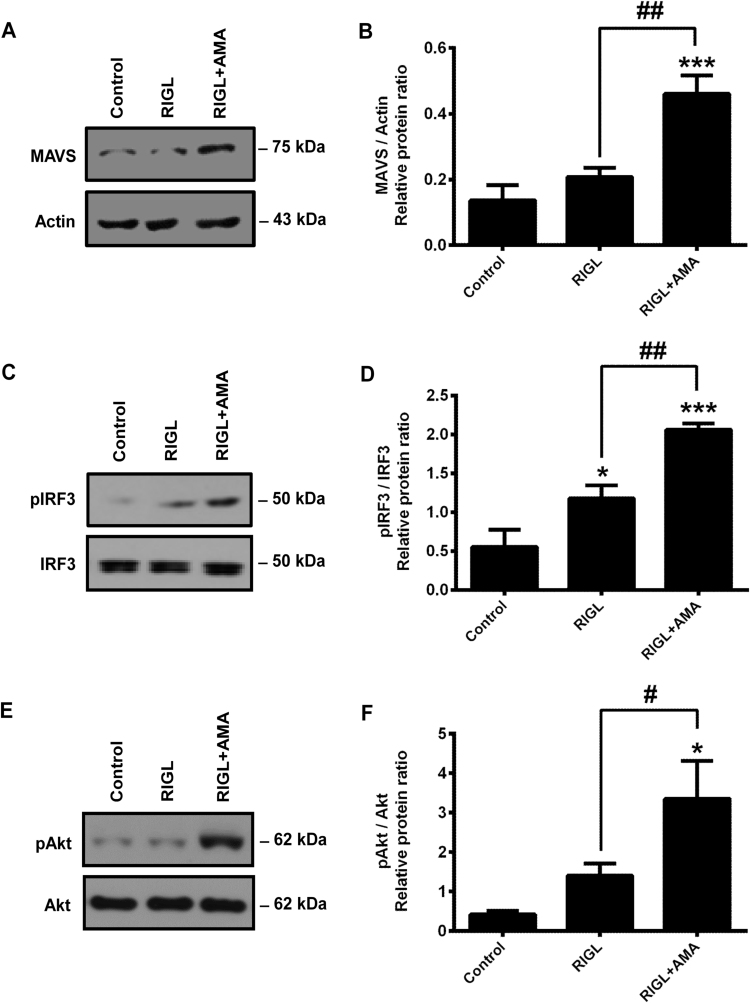
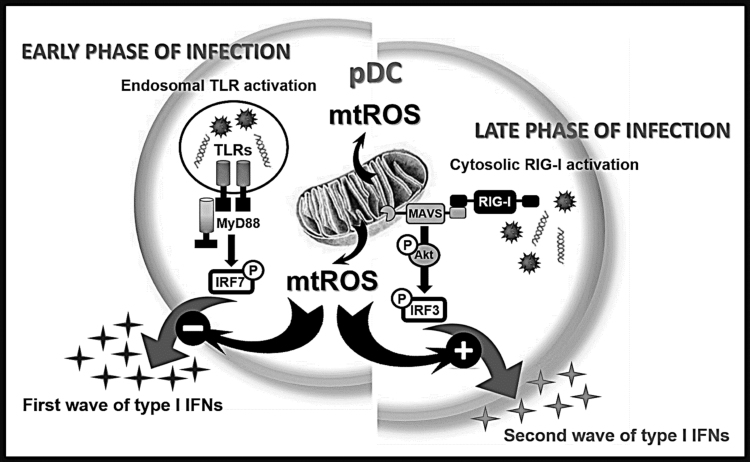
Mitochondrial reactive oxygen species (mtROS) generated continuously under physiological conditions have recently emerged as critical players in the regulation of immune signaling pathways. In this study we have investigated the regulation of antiviral signaling by increased mtROS production in plasmacytoid dendritic cells (pDCs), which, as major producers of type I interferons (IFN), are the key coordinators of antiviral immunity. The early phase of type I IFN production in pDCs is mediated by endosomal Toll-like receptors (TLRs), whereas the late phase of IFN response can also be triggered by cytosolic retinoic acid-inducible gene-I (RIG-I), expression of which is induced upon TLR stimulation. Therefore, pDCs provide an ideal model to study the impact of elevated mtROS on the antiviral signaling pathways initiated by receptors with distinct subcellular localization. We found that elevated level of mtROS alone did not change the phenotype and the baseline cytokine profile of resting pDCs. Nevertheless increased mtROS levels in pDCs lowered the TLR9-induced secretion of pro-inflammatory mediators slightly, whereas reduced type I IFN production markedly via blocking phosphorylation of interferon regulatory factor 7 (IRF7), the key transcription factor of the TLR9 signaling pathway. The TLR9-induced expression of RIG-I in pDCs was also negatively regulated by enhanced mtROS production. On the contrary, elevated mtROS significantly augmented the RIG-I-stimulated expression of type I IFNs, as well as the expression of mitochondrial antiviral-signaling (MAVS) protein and the phosphorylation of Akt and IRF3 that are essential components of RIG-I signaling. Collectively, our data suggest that increased mtROS exert diverse immunoregulatory functions in pDCs both in the early and late phase of type I IFN responses depending on which type of viral sensing pathway is stimulated.
Keywords: Antiviral response; Endosomal TLR signaling; Mitochondrial ROS; Plasmacytoid dendritic cell; RIG-I signaling; Type I interferon.
Copyright © 2017 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Regulatory NLRs Control the RLR-Mediated Type I Interferon and Inflammatory Responses in Human Dendritic Cells.Front Immunol. 2018 Oct 5;9:2314. doi: 10.3389/fimmu.2018.02314. eCollection 2018. Front Immunol. 2018. PMID: 30344524 Free PMC article.
-
Human Plasmacytoid and Monocyte-Derived Dendritic Cells Display Distinct Metabolic Profile Upon RIG-I Activation.Front Immunol. 2018 Dec 21;9:3070. doi: 10.3389/fimmu.2018.03070. eCollection 2018. Front Immunol. 2018. PMID: 30622542 Free PMC article.
-
TLR ligands upregulate RIG-I expression in human plasmacytoid dendritic cells in a type I IFN-independent manner.Immunol Cell Biol. 2014 Sep;92(8):671-8. doi: 10.1038/icb.2014.38. Epub 2014 May 20. Immunol Cell Biol. 2014. PMID: 24839978
-
Mechanisms and pathways of innate immune activation and regulation in health and cancer.Hum Vaccin Immunother. 2014;10(11):3270-85. doi: 10.4161/21645515.2014.979640. Hum Vaccin Immunother. 2014. PMID: 25625930 Free PMC article. Review.
-
Plasmacytoid dendritic cells in antiviral immunity and autoimmunity.Sci China Life Sci. 2010 Feb;53(2):172-82. doi: 10.1007/s11427-010-0045-0. Epub 2010 Mar 7. Sci China Life Sci. 2010. PMID: 20596824 Free PMC article. Review.
Cited by
-
Conceptus-modulated innate immune function during early pregnancy in ruminants: a review.Anim Reprod. 2021 May 10;18(1):e20200048. doi: 10.1590/1984-3143-AR2020-0048. Anim Reprod. 2021. PMID: 34122650 Free PMC article. Review.
-
Regulatory NLRs Control the RLR-Mediated Type I Interferon and Inflammatory Responses in Human Dendritic Cells.Front Immunol. 2018 Oct 5;9:2314. doi: 10.3389/fimmu.2018.02314. eCollection 2018. Front Immunol. 2018. PMID: 30344524 Free PMC article.
-
Therapeutic Targeting of Endosome and Mitochondrial Reactive Oxygen Species Protects Mice From Influenza Virus Morbidity.Front Pharmacol. 2022 Mar 23;13:870156. doi: 10.3389/fphar.2022.870156. eCollection 2022. Front Pharmacol. 2022. PMID: 35401240 Free PMC article.
-
Type I Interferon Production of Plasmacytoid Dendritic Cells under Control.Int J Mol Sci. 2021 Apr 18;22(8):4190. doi: 10.3390/ijms22084190. Int J Mol Sci. 2021. PMID: 33919546 Free PMC article. Review.
-
Oxidative Stress Attenuates TLR3 Responsiveness and Impairs Anti-viral Mechanisms in Bronchial Epithelial Cells From COPD and Asthma Patients.Front Immunol. 2019 Nov 29;10:2765. doi: 10.3389/fimmu.2019.02765. eCollection 2019. Front Immunol. 2019. PMID: 31849956 Free PMC article.
References
-
- Muller F.L., Liu Y., Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004;279(47):49064–49073. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
