

Mitochondrial energetics in the kidney
- PMID: 28804120
- PMCID: PMC5965678
- DOI: 10.1038/nrneph.2017.107
Mitochondrial energetics in the kidney
Abstract
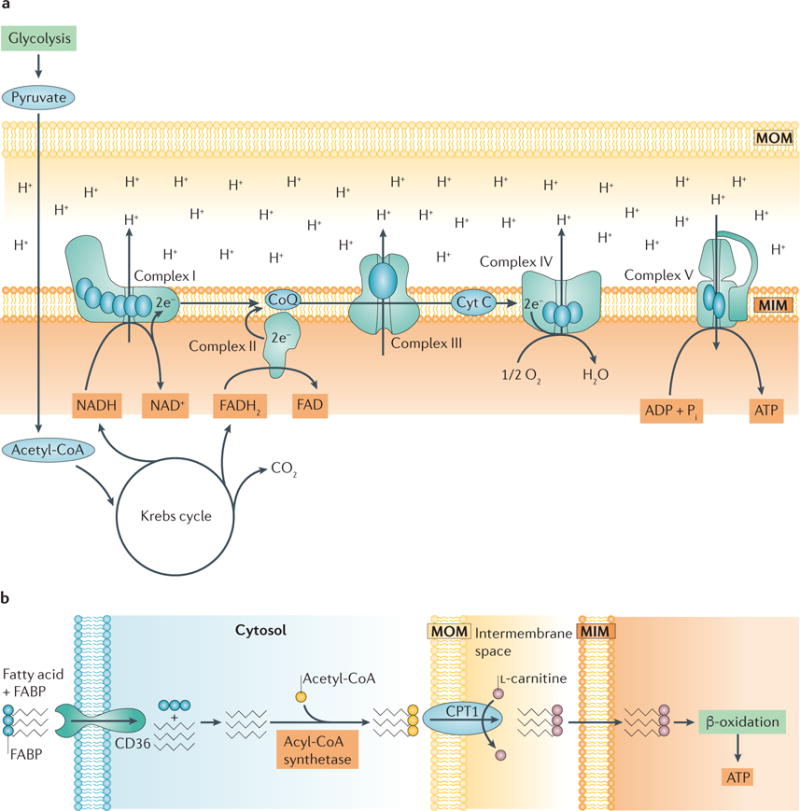
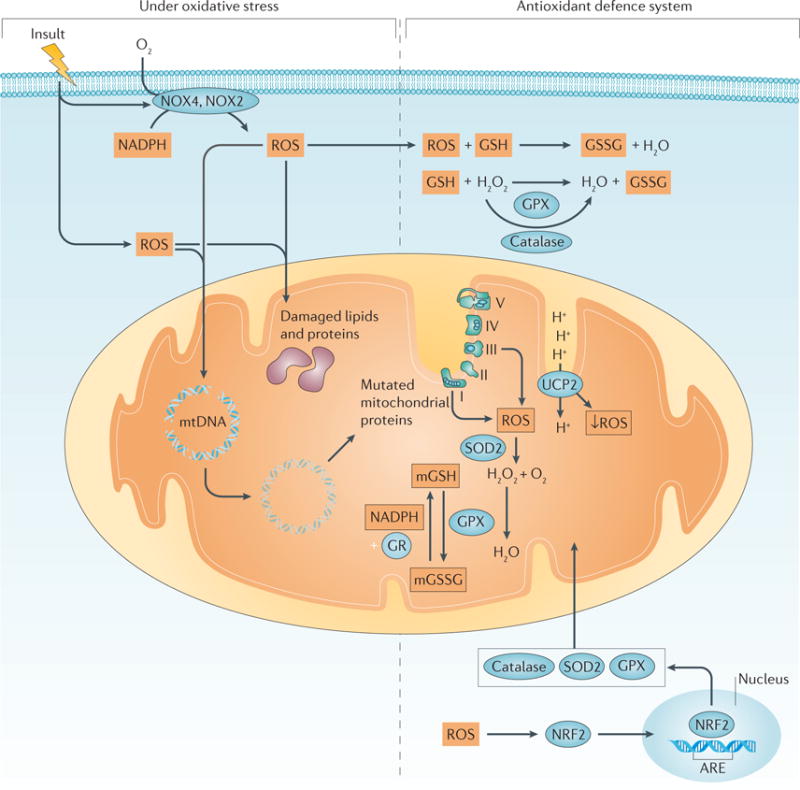
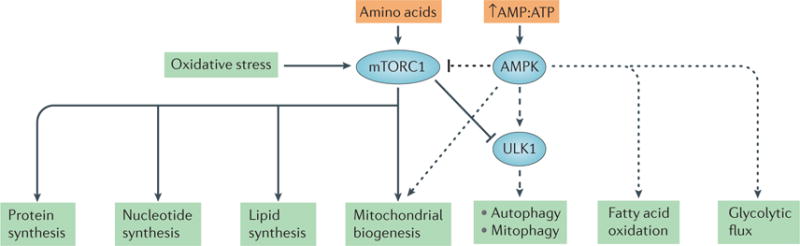
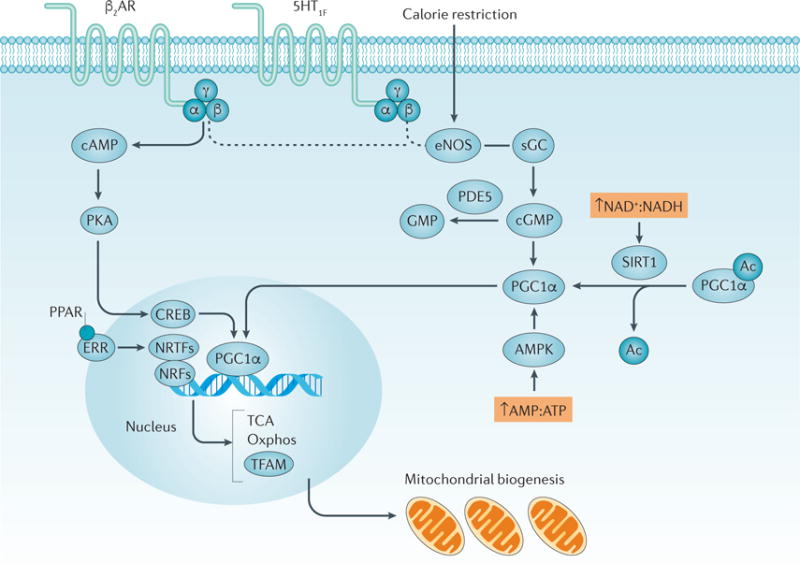
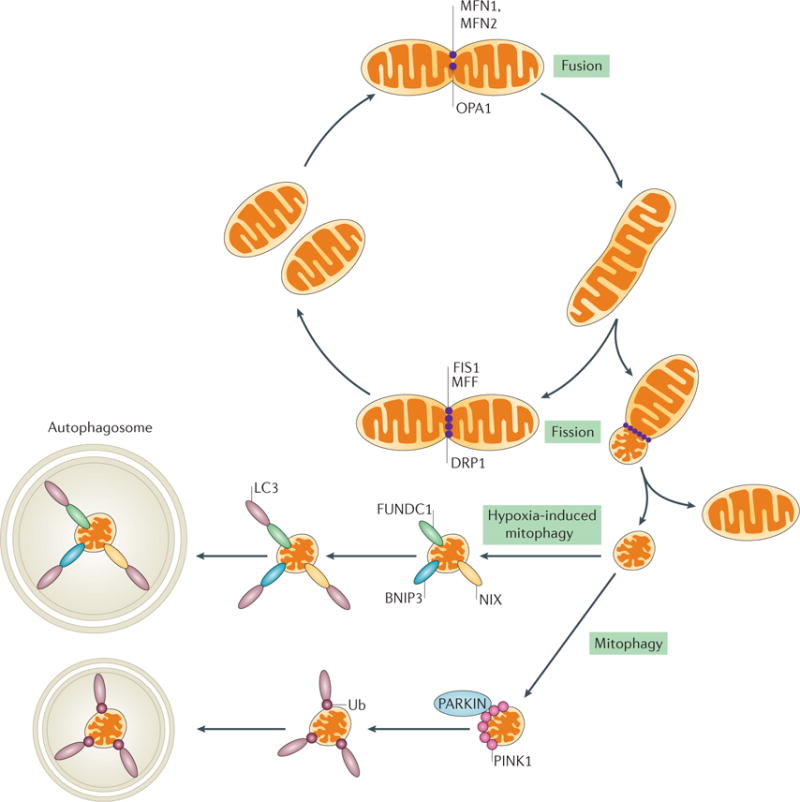
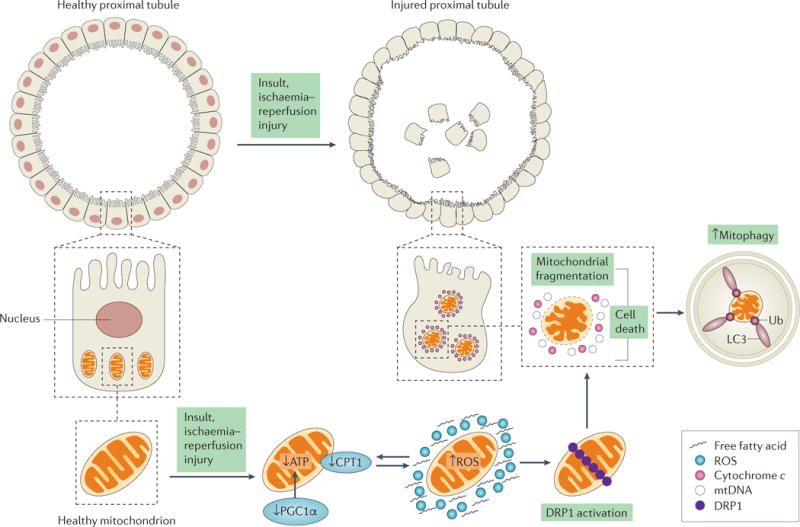
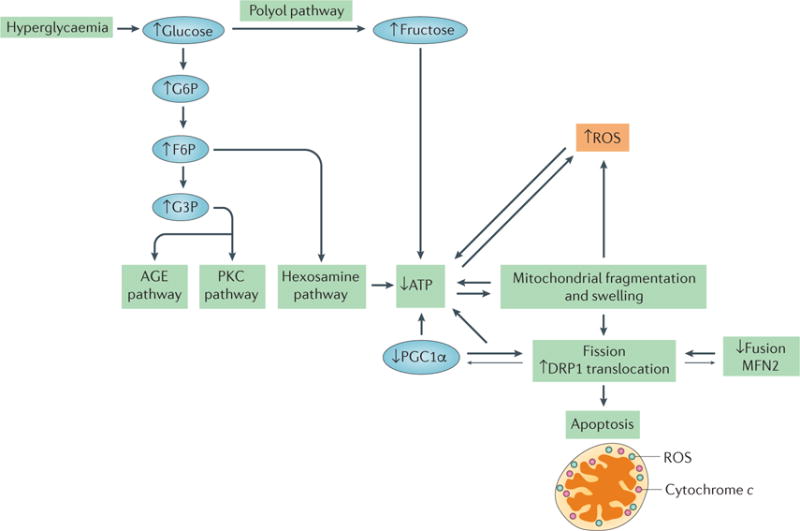
The kidney requires a large number of mitochondria to remove waste from the blood and regulate fluid and electrolyte balance. Mitochondria provide the energy to drive these important functions and can adapt to different metabolic conditions through a number of signalling pathways (for example, mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) pathways) that activate the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α), and by balancing mitochondrial dynamics and energetics to maintain mitochondrial homeostasis. Mitochondrial dysfunction leads to a decrease in ATP production, alterations in cellular functions and structure, and the loss of renal function. Persistent mitochondrial dysfunction has a role in the early stages and progression of renal diseases, such as acute kidney injury (AKI) and diabetic nephropathy, as it disrupts mitochondrial homeostasis and thus normal kidney function. Improving mitochondrial homeostasis and function has the potential to restore renal function, and administering compounds that stimulate mitochondrial biogenesis can restore mitochondrial and renal function in mouse models of AKI and diabetes mellitus. Furthermore, inhibiting the fission protein dynamin 1-like protein (DRP1) might ameliorate ischaemic renal injury by blocking mitochondrial fission.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Mitochondrial biogenesis: pharmacological approaches.Curr Pharm Des. 2014;20(35):5507-9. doi: 10.2174/138161282035140911142118. Curr Pharm Des. 2014. PMID: 24606795
-
Melatonin attenuates renal fibrosis in diabetic mice by activating the AMPK/PGC1α signaling pathway and rescuing mitochondrial function.Mol Med Rep. 2019 Feb;19(2):1318-1330. doi: 10.3892/mmr.2018.9708. Epub 2018 Nov 29. Mol Med Rep. 2019. PMID: 30535482
-
The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases.Biomolecules. 2020 Feb 24;10(2):347. doi: 10.3390/biom10020347. Biomolecules. 2020. PMID: 32102312 Free PMC article. Review.
-
Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes.Acta Med Okayama. 2016 Jun;70(3):151-8. doi: 10.18926/AMO/54413. Acta Med Okayama. 2016. PMID: 27339203 Review.
-
Klotho ameliorates diabetic nephropathy via LKB1-AMPK-PGC1α-mediated renal mitochondrial protection.Biochem Biophys Res Commun. 2021 Jan 1;534:1040-1046. doi: 10.1016/j.bbrc.2020.10.040. Epub 2020 Oct 26. Biochem Biophys Res Commun. 2021. PMID: 33121684
Cited by
-
Efficient fluorescent recognition of ATP/GTP by a water-soluble bisquinolinium pyridine-2,6-dicarboxamide compound. Crystal structures, spectroscopic studies and interaction mode with DNA.RSC Adv. 2022 Sep 29;12(43):27826-27838. doi: 10.1039/d2ra05040d. eCollection 2022 Sep 28. RSC Adv. 2022. PMID: 36320280 Free PMC article.
-
Diverse Roles of Mitochondria in Renal Injury from Environmental Toxicants and Therapeutic Drugs.Int J Mol Sci. 2021 Apr 17;22(8):4172. doi: 10.3390/ijms22084172. Int J Mol Sci. 2021. PMID: 33920653 Free PMC article. Review.
-
Sodium butyrate improves mitochondrial function and kidney tissue injury in diabetic kidney disease via the AMPK/PGC-1α pathway.Ren Fail. 2023;45(2):2287129. doi: 10.1080/0886022X.2023.2287129. Epub 2023 Dec 10. Ren Fail. 2023. PMID: 38073119 Free PMC article.
-
Extracellular vesicle-encapsulated IL-10 as novel nanotherapeutics against ischemic AKI.Sci Adv. 2020 Aug 12;6(33):eaaz0748. doi: 10.1126/sciadv.aaz0748. eCollection 2020 Aug. Sci Adv. 2020. PMID: 32851154 Free PMC article.
-
Molecular Mechanisms of AKI in the Elderly: From Animal Models to Therapeutic Intervention.J Clin Med. 2020 Aug 8;9(8):2574. doi: 10.3390/jcm9082574. J Clin Med. 2020. PMID: 32784471 Free PMC article. Review.
References
-
- O’Connor PM. Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol. 2006;33:961–967. - PubMed
-
- Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol. 1986;48:9–31. - PubMed
-
- Holechek MJ, et al. Glomerular filtration: an overview. Nephrol Nurs J. 2003;30:285–290. quiz 281–282. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
