

Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later
- PMID: 28758305
- PMCID: PMC5539911
- DOI: 10.1111/prd.12181
Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later
Abstract
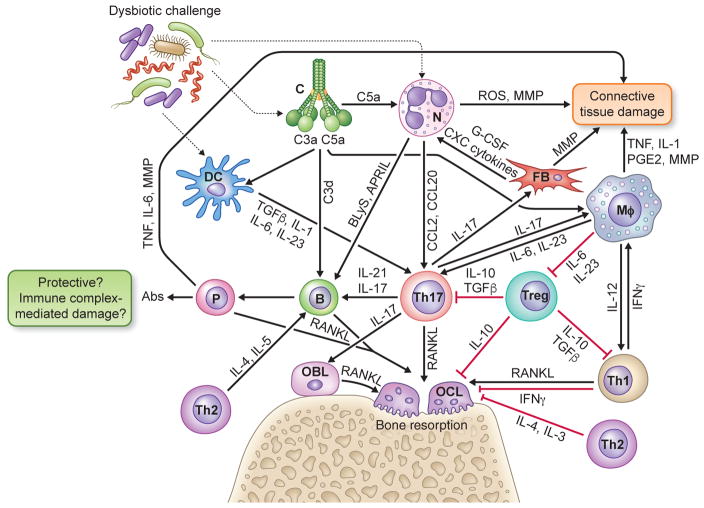
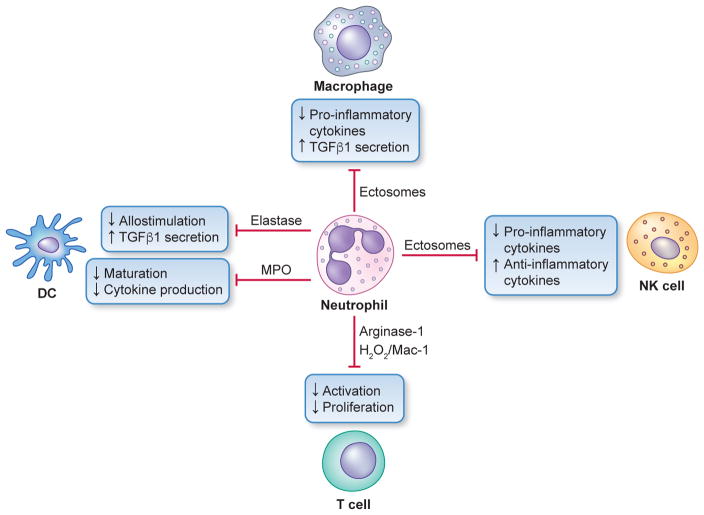
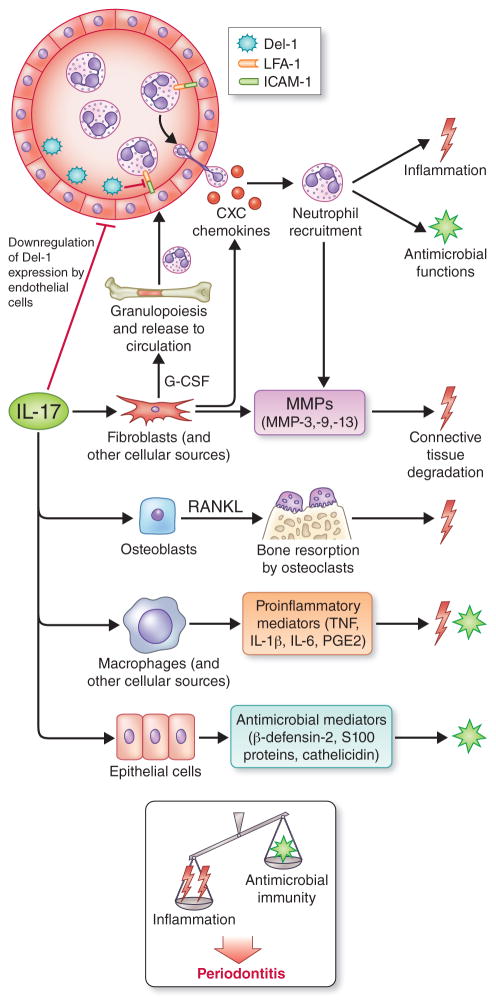
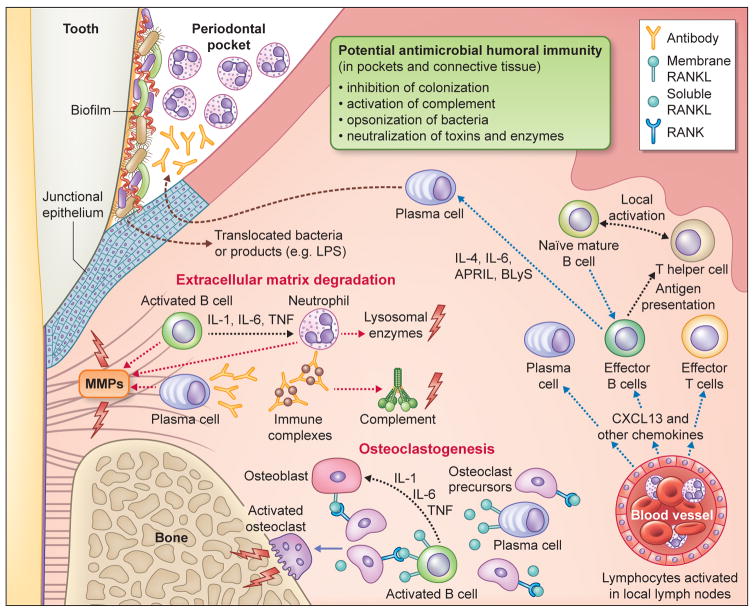
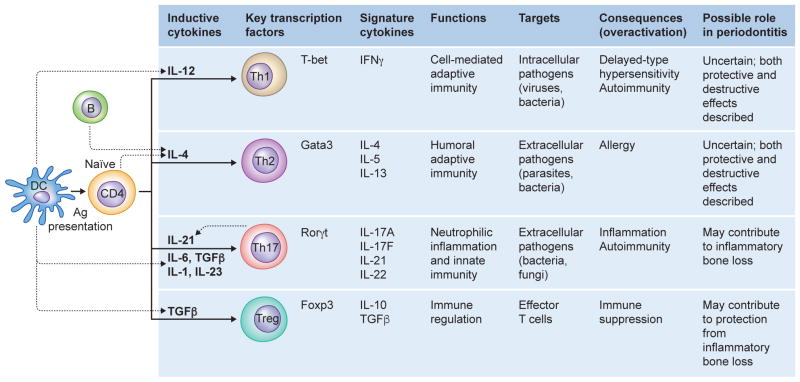
In their classic 1976 paper, Page & Schroeder described the histopathologic events and the types of myeloid cells and lymphocytes involved in the initiation and progression of inflammatory periodontal disease. The staging of periodontal disease pathogenesis as 'initial', 'early', 'established' and 'advanced' lesions productively guided subsequent research in the field and remains fundamentally valid. However, major advances regarding the cellular and molecular mechanisms underlying the induction, regulation and effector functions of immune and inflammatory responses necessitate a reassessment of their work and its integration with emerging new concepts. We now know that each type of leukocyte is actually represented by functionally distinct subsets with different, or even conflicting, roles in immunity and inflammation. Unexpectedly, neutrophils, traditionally regarded as merely antimicrobial effectors in acute conditions and protagonists of the 'initial' lesion, are currently appreciated for their functional versatility and critical roles in chronic inflammation. Moreover, an entirely new field of study, osteoimmunology, has emerged and sheds light on the impact of immunoinflammatory events on the skeletal system. These developments and the molecular dissection of crosstalk interactions between innate and adaptive leukocytes, as well as between the immune system and local homeostatic mechanisms, offer a more nuanced understanding of the host response in periodontitis, with profound implications for treatment. At the same time, deeper insights have generated new questions, many of which remain unanswered. In this review, 40 years after Page & Schroeder proposed their model, we summarize enduring and emerging advances in periodontal disease pathogenesis.
© 2017 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Figures
Similar articles
-
An update on periodontal aetiopathogenesis and clinical implications.Ann R Australas Coll Dent Surg. 2008 Jun;19:96-101. Ann R Australas Coll Dent Surg. 2008. PMID: 22073461
-
Molecular aspects of the pathogenesis of periodontitis.Periodontol 2000. 2015 Oct;69(1):7-17. doi: 10.1111/prd.12104. Periodontol 2000. 2015. PMID: 26252398
-
Immune and regulatory functions of neutrophils in inflammatory bone loss.Semin Immunol. 2016 Apr;28(2):146-58. doi: 10.1016/j.smim.2016.02.002. Epub 2016 Feb 28. Semin Immunol. 2016. PMID: 26936034 Free PMC article. Review.
-
Changing periodontal paradigms: therapeutic implications.Int J Periodontics Restorative Dent. 2000 Aug;20(4):336-57. Int J Periodontics Restorative Dent. 2000. PMID: 11203574 Review.
-
Cytokine responses against periodontal infection: protective and destructive roles.Periodontol 2000. 2010 Feb;52(1):163-206. doi: 10.1111/j.1600-0757.2009.00321.x. Periodontol 2000. 2010. PMID: 20017801 Review. No abstract available.
Cited by
-
Neutrophils in the periodontium: Interactions with pathogens and roles in tissue homeostasis and inflammation.Immunol Rev. 2023 Mar;314(1):93-110. doi: 10.1111/imr.13152. Epub 2022 Oct 22. Immunol Rev. 2023. PMID: 36271881 Free PMC article. Review.
-
Models of periodontal disease pathogenesis: A journey through time.J Indian Soc Periodontol. 2022 May-Jun;26(3):204-212. doi: 10.4103/jisp.jisp_294_21. Epub 2022 May 2. J Indian Soc Periodontol. 2022. PMID: 35602539 Free PMC article. Review.
-
A20 Restricts Inflammatory Response and Desensitizes Gingival Keratinocytes to Apoptosis.Front Immunol. 2020 Mar 10;11:365. doi: 10.3389/fimmu.2020.00365. eCollection 2020. Front Immunol. 2020. PMID: 32218782 Free PMC article.
-
Regulatory T Lymphocytes in Periodontitis: A Translational View.Mediators Inflamm. 2018 Apr 2;2018:7806912. doi: 10.1155/2018/7806912. eCollection 2018. Mediators Inflamm. 2018. PMID: 29805313 Free PMC article. Review.
-
Clonal hematopoiesis driven by mutated DNMT3A promotes inflammatory bone loss.Cell. 2024 Jul 11;187(14):3690-3711.e19. doi: 10.1016/j.cell.2024.05.003. Epub 2024 Jun 4. Cell. 2024. PMID: 38838669 Free PMC article.
References
-
- Abe T, AlSarhan M, Benakanakere MR, Maekawa T, Kinane DF, Cancro MP, Korostoff JM, Hajishengallis G. The B cell-stimulatory cytokines BLyS and APRIL are elevated in human periodontitis and are required for B cell-dependent bone loss in experimental murine periodontitis. J Immunol. 2015;195:1427–1435. - PMC - PubMed
-
- Allam JP, Duan Y, Heinemann F, Winter J, Gotz W, Deschner J, Wenghoefer M, Bieber T, Jepsen S, Novak N. IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J Clin Periodontol. 2011;38:879–886. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
