

Neonatal Retinoblastoma
- PMID: 28695165
- PMCID: PMC5473090
- DOI: 10.4103/apjon.apjon_18_17
Neonatal Retinoblastoma
Abstract
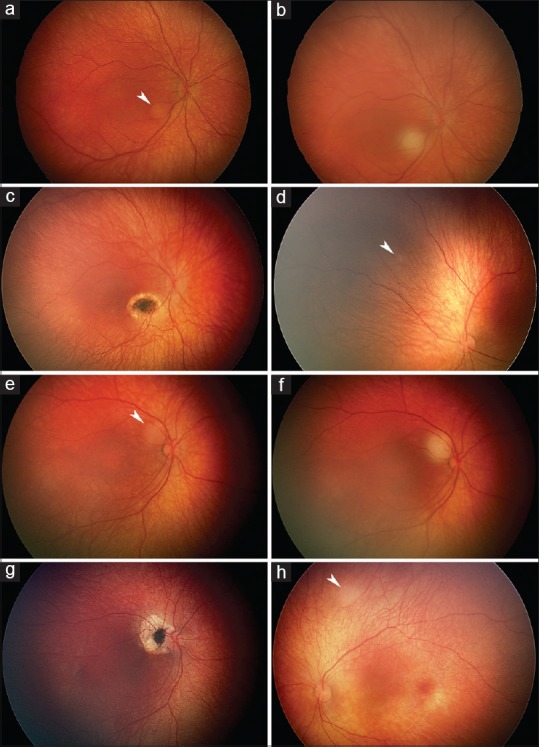
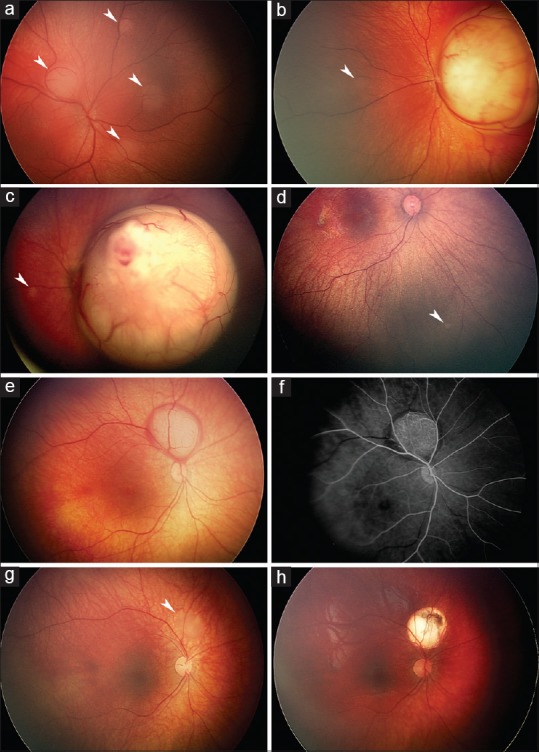
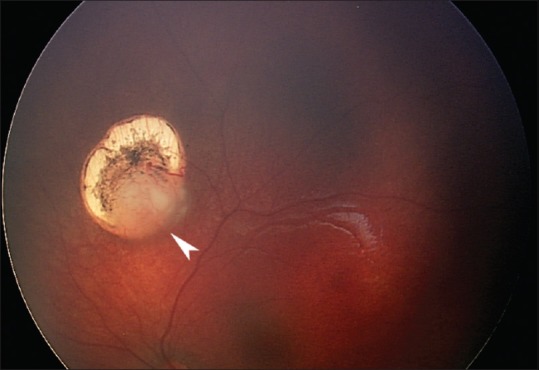
From 7% to 10% of all retinoblastomas and from 44% to 71% of familial retinoblastomas in developed countries are diagnosed in the neonatal period, usually through pre- or post-natal screening prompted by a positive family history and sometimes serendipitously during screening for retinopathy of prematurity or other reasons. In developing countries, neonatal diagnosis of retinoblastoma has been less common. Neonatal retinoblastoma generally develops from a germline mutation of RB1, the retinoblastoma gene, even when the family history is negative and is thus usually hereditary. At least one-half of infants with neonatal retinoblastoma have unilateral tumors when the diagnosis is made, typically the International Intraocular Retinoblastoma Classification (Murphree) Group B or higher, but most germline mutation carriers will progress to bilateral involvement, typically Group A in the fellow eye. Neonatal leukokoria usually leads to the diagnosis in children without a family history of retinoblastoma, and a Group C tumor or higher is typical in the more advanced involved eye. Almost all infants with neonatal retinoblastoma have at least one eye with a tumor in proximity to the foveola, but the macula of the fellow eye is frequently spared. Consequently, loss of reading vision from both eyes is exceptional. A primary ectopic intracranial neuroblastic tumor known as trilateral retinoblastoma is no more common after neonatal than other retinoblastoma. For many reasons, neonatal retinoblastoma may be a challenge to eradicate, and the early age at diagnosis and relatively small tumors do not guarantee the preservation of both eyes of every involved child. Oncology nurses can be instrumental in contributing to better outcomes by ensuring that hereditary retinoblastoma survivors receive genetic counseling, by referring families of survivors to early screening programs when they are planning for a baby, and by providing psychological and practical support for parents when neonatal retinoblastoma has been diagnosed.
Keywords: Germline mutation; neonatal cancer; prenatal diagnosis; retinoblastoma.
Conflict of interest statement
There are no conflicts of interest.
Figures
Similar articles
-
Prenatal versus Postnatal Screening for Familial Retinoblastoma.Ophthalmology. 2016 Dec;123(12):2610-2617. doi: 10.1016/j.ophtha.2016.08.027. Epub 2016 Oct 3. Ophthalmology. 2016. PMID: 27712844
-
Correlation between Family RB1 Gene Pathogenic Variant with Clinical Features and Prognosis of Retinoblastoma under 5 Years Old.Dis Markers. 2021 Jul 12;2021:9981028. doi: 10.1155/2021/9981028. eCollection 2021. Dis Markers. 2021. PMID: 34336010 Free PMC article.
-
Prenatal diagnosis of bilateral retinoblastomas by multimodality fetal imaging: case report and review of the literature.Clin Imaging. 2021 Oct;78:121-126. doi: 10.1016/j.clinimag.2021.03.023. Epub 2021 Mar 20. Clin Imaging. 2021. PMID: 33774578 Review.
-
RB1 gene mutations in Argentine retinoblastoma patients. Implications for genetic counseling.PLoS One. 2017 Dec 20;12(12):e0189736. doi: 10.1371/journal.pone.0189736. eCollection 2017. PLoS One. 2017. PMID: 29261756 Free PMC article.
-
[Retinoblastoma: recent advances].Bull Cancer. 2014 Apr;101(4):380-7. doi: 10.1684/bdc.2014.1931. Bull Cancer. 2014. PMID: 24793631 Review. French.
Cited by
-
The clinical diagnostic value of plasma miR-592 and miR-217-3p levels in retinoblastoma.J Med Biochem. 2022 Oct 15;41(4):497-505. doi: 10.5937/jomb0-34794. J Med Biochem. 2022. PMID: 36381083 Free PMC article.
-
Dissecting the Transcriptional and Chromatin Accessibility Heterogeneity of Proliferating Cone Precursors in Human Retinoblastoma Tumors by Single Cell Sequencing-Opening Pathways to New Therapeutic Strategies?Invest Ophthalmol Vis Sci. 2021 May 3;62(6):18. doi: 10.1167/iovs.62.6.18. Invest Ophthalmol Vis Sci. 2021. PMID: 34003213 Free PMC article.
-
lncRNA FEZF1‑AS1 promotes migration, invasion and epithelial‑mesenchymal transition of retinoblastoma cells by targeting miR‑1236‑3p.Mol Med Rep. 2020 Nov;22(5):3635-3644. doi: 10.3892/mmr.2020.11478. Epub 2020 Sep 2. Mol Med Rep. 2020. PMID: 32901841 Free PMC article.
-
Spontaneous regression of an isolated retinal astrocytic hamartoma in a newborn: a case report.BMC Ophthalmol. 2023 Sep 26;23(1):395. doi: 10.1186/s12886-023-03135-5. BMC Ophthalmol. 2023. PMID: 37752483 Free PMC article.
-
Survival and ocular preservation in a long-term cohort of Japanese patients with retinoblastoma.BMC Pediatr. 2020 Jan 28;20(1):37. doi: 10.1186/s12887-020-1923-7. BMC Pediatr. 2020. PMID: 31992242 Free PMC article.
References
-
- Halperin EC. Neonatal neoplasms. Int J Radiat Oncol Biol Phys. 2000;47:171–8. - PubMed
-
- Moore SW, Satgé D, Sasco AJ, Zimmermann A, Plaschkes J. The epidemiology of neonatal tumours. Report of an international working group. Pediatr Surg Int. 2003;19:509–19. - PubMed
-
- Abramson DH, Du TT, Beaverson KL. (Neonatal) retinoblastoma in the first month of life. Arch Ophthalmol. 2002;120:738–42. - PubMed
-
- Xue H, Horwitz JR, Smith MB, Lally KP, Black CT, Cangir A, et al. Malignant solid tumors in neonates: A 40-year review. J Pediatr Surg. 1995;30:543–5. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
