

Cross talk between progesterone receptors and retinoic acid receptors in regulation of cytokeratin 5-positive breast cancer cells
- PMID: 28692043
- PMCID: PMC5668194
- DOI: 10.1038/onc.2017.204
Cross talk between progesterone receptors and retinoic acid receptors in regulation of cytokeratin 5-positive breast cancer cells
Abstract
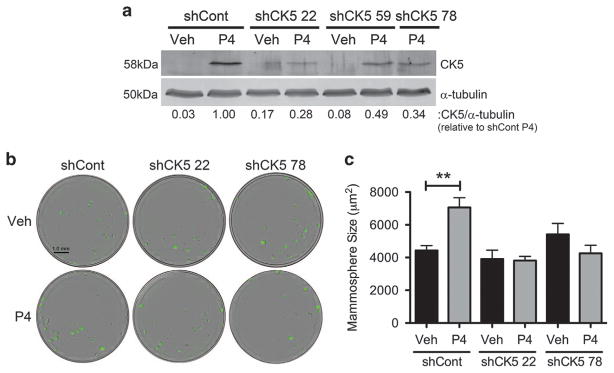
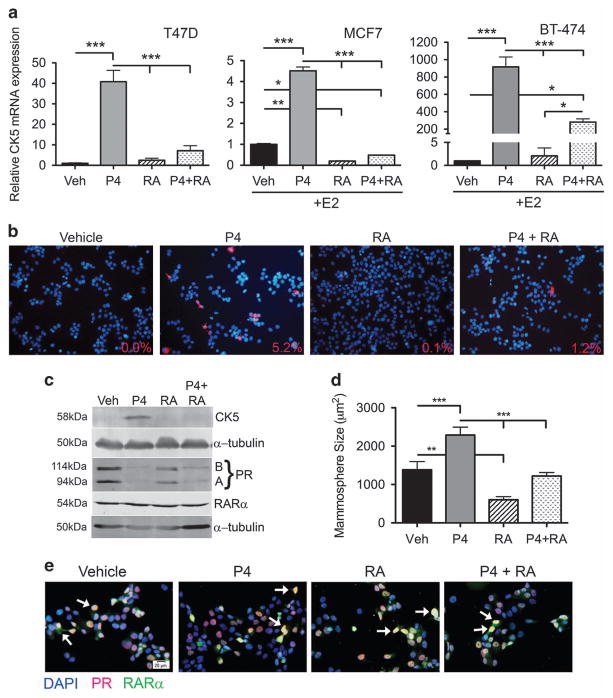
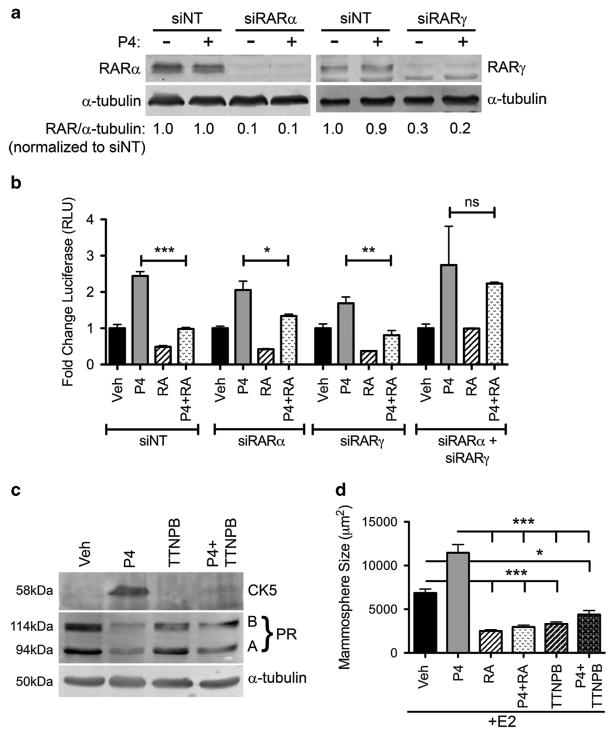
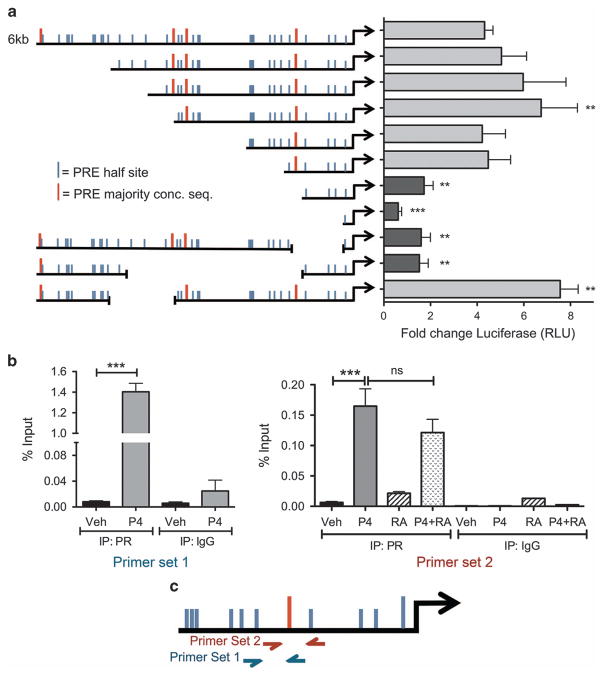
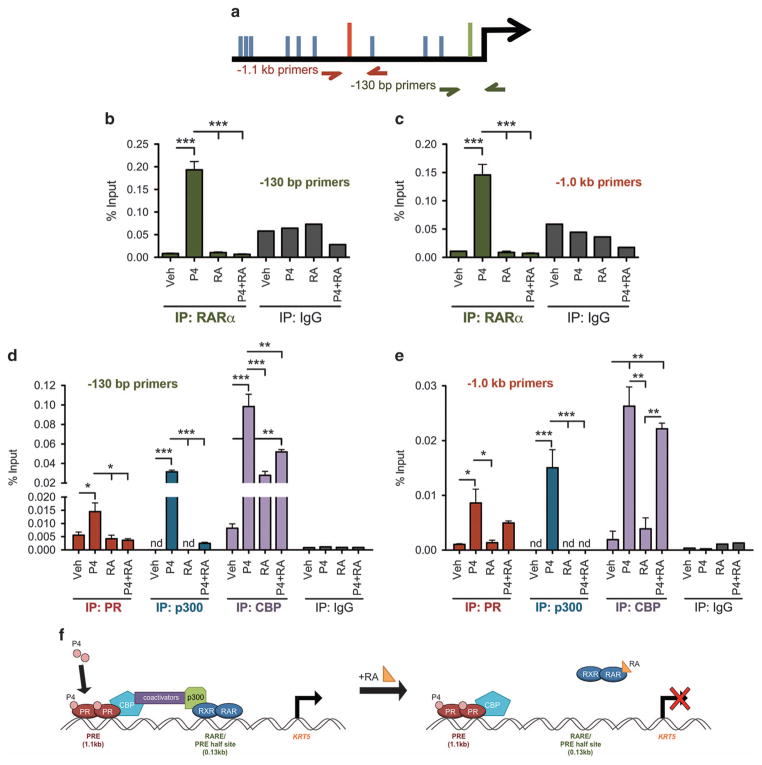
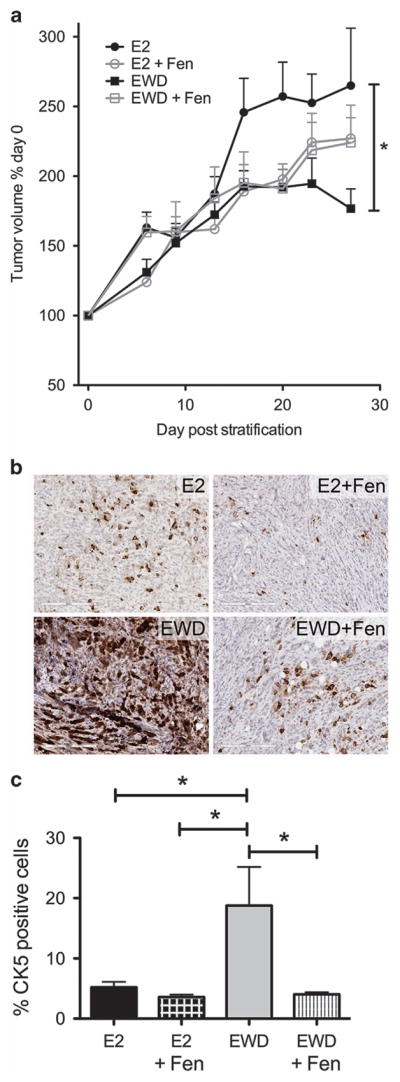
Half of estrogen receptor-positive breast cancers contain a subpopulation of cytokeratin 5 (CK5)-expressing cells that are therapy resistant and exhibit increased cancer stem cell (CSC) properties. We and others have demonstrated that progesterone (P4) increases CK5+ breast cancer cells. We previously discovered that retinoids block P4 induction of CK5+ cells. Here we investigated the mechanisms by which progesterone receptors (PR) and retinoic acid receptors (RAR) regulate CK5 expression and breast CSC activity. After P4 treatment, sorted CK5+ compared to CK5- cells were more tumorigenic in vivo. In vitro, P4-treated breast cancer cells formed larger mammospheres and silencing of CK5 using small hairpin RNA abolished this P4-dependent increase in mammosphere size. Retinoic acid (RA) treatment blocked the P4 increase in CK5+ cells and prevented the P4 increase in mammosphere size. Dual small interfering RNA (siRNA) silencing of RARα and RARγ reversed RA blockade of P4-induced CK5. Using promoter deletion analysis, we identified a region 1.1 kb upstream of the CK5 transcriptional start site that is necessary for P4 activation and contains a putative progesterone response element (PRE). We confirmed by chromatin immunoprecipitation that P4 recruits PR to the CK5 promoter near the -1.1 kb essential PRE, and also to a proximal region near -130 bp that contains PRE half-sites and a RA response element (RARE). RA induced loss of PR binding only at the proximal site. Interestingly, RARα was recruited to the -1.1 kb PRE and the -130 bp PRE/RARE regions with P4, but not RA alone or RA plus P4. Treatment of breast cancer xenografts in vivo with the retinoid fenretinide reduced the accumulation of CK5+ cells during estrogen depletion. This reduction, together with the inhibition of CK5+ cell expansion through RAR/PR cross talk, may explain the efficacy of retinoids in prevention of some breast cancer recurrences.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Progesterone-inducible cytokeratin 5-positive cells in luminal breast cancer exhibit progenitor properties.Horm Cancer. 2013 Feb;4(1):36-49. doi: 10.1007/s12672-012-0127-5. Epub 2012 Nov 27. Horm Cancer. 2013. PMID: 23184698 Free PMC article.
-
Progesterone downregulation of miR-141 contributes to expansion of stem-like breast cancer cells through maintenance of progesterone receptor and Stat5a.Oncogene. 2015 Jul;34(28):3676-87. doi: 10.1038/onc.2014.298. Epub 2014 Sep 22. Oncogene. 2015. PMID: 25241899 Free PMC article.
-
A high-content assay to identify small-molecule modulators of a cancer stem cell population in luminal breast cancer.J Biomol Screen. 2012 Oct;17(9):1211-20. doi: 10.1177/1087057112452138. Epub 2012 Jun 29. J Biomol Screen. 2012. PMID: 22751729 Free PMC article.
-
Steroid hormones, steroid receptors, and breast cancer stem cells.J Mammary Gland Biol Neoplasia. 2015 Jun;20(1-2):39-50. doi: 10.1007/s10911-015-9340-5. Epub 2015 Aug 12. J Mammary Gland Biol Neoplasia. 2015. PMID: 26265122 Free PMC article. Review.
-
Progesterone receptor-estrogen receptor crosstalk: a novel insight.Trends Endocrinol Metab. 2015 Sep;26(9):453-4. doi: 10.1016/j.tem.2015.08.002. Epub 2015 Aug 12. Trends Endocrinol Metab. 2015. PMID: 26277479 Review.
Cited by
-
Impaired redox regulation of estrogen metabolizing proteins is important determinant of human breast cancers.Cancer Cell Int. 2019 May 15;19:111. doi: 10.1186/s12935-019-0826-x. eCollection 2019. Cancer Cell Int. 2019. PMID: 31114446 Free PMC article. Review.
-
90 YEARS OF PROGESTERONE: Progesterone and progesterone receptors in breast cancer: past, present, future.J Mol Endocrinol. 2020 Jul;65(1):T49-T63. doi: 10.1530/JME-20-0104. J Mol Endocrinol. 2020. PMID: 32485679 Free PMC article. Review.
-
Cytokeratin 5 alters β-catenin dynamics in breast cancer cells.Oncogene. 2020 Mar;39(12):2478-2492. doi: 10.1038/s41388-020-1164-0. Epub 2020 Jan 27. Oncogene. 2020. PMID: 31988452 Free PMC article.
-
Breast Cancer Suppression by Progesterone Receptors Is Mediated by Their Modulation of Estrogen Receptors and RNA Polymerase III.Cancer Res. 2017 Sep 15;77(18):4934-4946. doi: 10.1158/0008-5472.CAN-16-3541. Epub 2017 Jul 20. Cancer Res. 2017. PMID: 28729413 Free PMC article.
-
Effects of polyclonal activators on cell differentiation and cytokine production of cultured invasive breast carcinoma of no special type, their association with tumour histopathological parameters and lymph node metastasis.Int J Immunopathol Pharmacol. 2020 Jan-Dec;34:2058738420950580. doi: 10.1177/2058738420950580. Int J Immunopathol Pharmacol. 2020. PMID: 33100082 Free PMC article.
References
-
- Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006;26:87–98. - PubMed
-
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
