

Review
doi: 10.1002/mas.21540.
Epub 2017 Jul 9.
The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics
Affiliations
- PMID: 28691345
- PMCID: PMC5799042
- DOI: 10.1002/mas.21540
Item in Clipboard
Review
The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics
Mass Spectrom Rev.
2020 May.
Abstract
Skyline is a freely available, open-source Windows client application for accelerating targeted proteomics experimentation, with an emphasis on the proteomics and mass spectrometry community as users and as contributors. This review covers the informatics encompassed by the Skyline ecosystem, from computationally assisted targeted mass spectrometry method development, to raw acquisition file data processing, and quantitative analysis and results sharing.
Keywords: informatics; quantitative mass spectrometry; targeted proteomics.
© 2017 Wiley Periodicals, Inc.
Figures
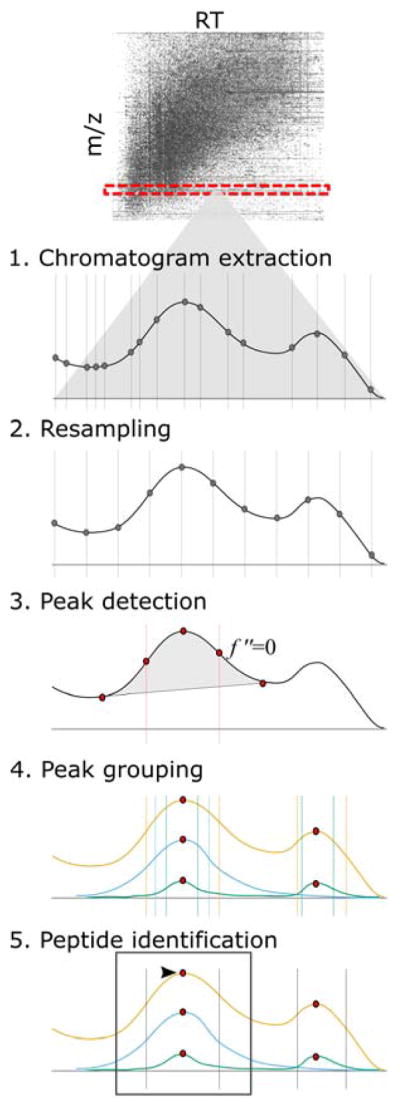
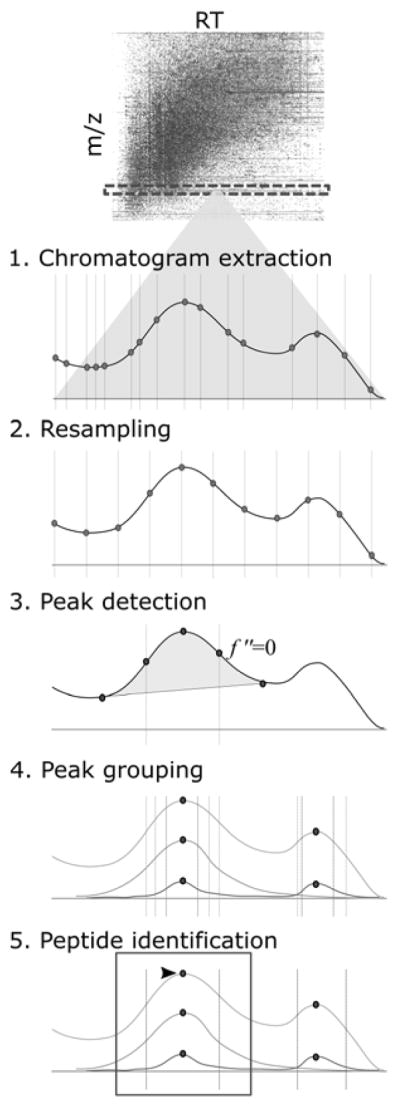
Six main steps are outlined, beginning with the development of a hypothesis and continuing through additional analyses, with examples of the associated Skyline ecosystem features.
Skyline derives information from native, vendor-specific file formats or from portable files, producing peak area calculations, and visualizations of the data.
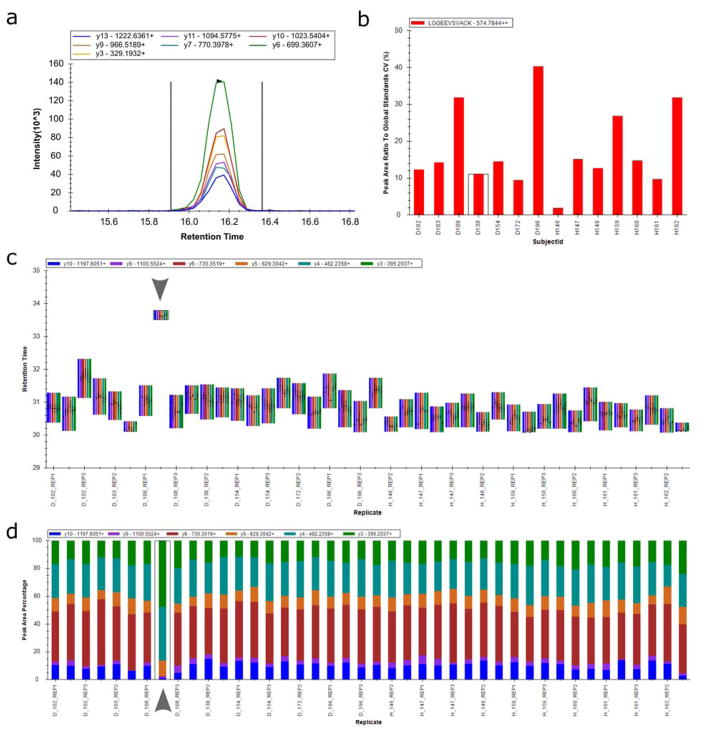
(a) Skyline chromatogram visualizations show the intensity at each resampled retention time point for all fragment ions (displayed as different colored lines identified in the legend) of a precursor, enabling researchers to assess Skyline’s automated peak picking or adjust integration boundaries if necessary. (b) Calculation of coefficient of variation (CV) informs researchers of the reproducibility of peptide peak areas (shown here as the peak area ratio to a global standard) over multiple acquisitions or custom-annotated groups of acquisitions. (c) Real-time updating visualization of precursor retention time across acquisitions enables quick identification of mis-picked peaks over many MS acquisition runs. Out of 42 replicates, the peptide shown here appears to elute three minutes late in one replicate (eighth from the left, marked with arrow) compared to all other replicates, an observation that may prompt the researcher to evaluate that picked peak in the chromatogram visualization pane. (d) Peak area is displayed here as the percentage contributed by each fragment ion of the precursor which allows the researcher to quickly evaluate data quality. For example, the boxed replicate (eighth run from the left, marked with arrow) displays a noticeably different distribution of contributed fragment peak areas, indicating that the picked peak group for this replicate may require further examination.
Similar articles
-
Skyline: an open source document editor for creating and analyzing targeted proteomics experiments.Bioinformatics. 2010 Apr 1;26(7):966-8. doi: 10.1093/bioinformatics/btq054. Epub 2010 Feb 9. Bioinformatics. 2010. PMID: 20147306 Free PMC article.
-
A framework for installable external tools in Skyline.Bioinformatics. 2014 Sep 1;30(17):2521-3. doi: 10.1093/bioinformatics/btu148. Epub 2014 May 9. Bioinformatics. 2014. PMID: 24813211 Free PMC article.
-
Panorama: a targeted proteomics knowledge base.J Proteome Res. 2014 Sep 5;13(9):4205-10. doi: 10.1021/pr5006636. Epub 2014 Aug 18. J Proteome Res. 2014. PMID: 25102069 Free PMC article.
-
Recent developments in proteome informatics for mass spectrometry analysis.Comb Chem High Throughput Screen. 2009 Feb;12(2):194-202. doi: 10.2174/138620709787315508. Comb Chem High Throughput Screen. 2009. PMID: 19199887 Review.
-
Proteomics by mass spectrometry: approaches, advances, and applications.Annu Rev Biomed Eng. 2009;11:49-79. doi: 10.1146/annurev-bioeng-061008-124934. Annu Rev Biomed Eng. 2009. PMID: 19400705 Review.
Cited by
-
Characterizing the flavodoxin landscape in Clostridioides difficile.Microbiol Spectr. 2024 Mar 5;12(3):e0189523. doi: 10.1128/spectrum.01895-23. Epub 2024 Feb 6. Microbiol Spectr. 2024. PMID: 38319052 Free PMC article.
-
Functional analysis of a common BAG3 allele associated with protection from heart failure.Nat Cardiovasc Res. 2023 Jul;2(7):615-628. doi: 10.1038/s44161-023-00288-w. Epub 2023 Jun 26. Nat Cardiovasc Res. 2023. PMID: 39195919
-
Substrate O-glycosylation actively regulates extracellular proteolysis.Protein Sci. 2024 Aug;33(8):e5128. doi: 10.1002/pro.5128. Protein Sci. 2024. PMID: 39074261 Free PMC article.
-
Rubisco packaging and stoichiometric composition of a native β-carboxysome.bioRxiv [Preprint]. 2024 Sep 21:2024.09.20.614183. doi: 10.1101/2024.09.20.614183. bioRxiv. 2024. Update in: Plant Physiol. 2024 Dec 24;197(1):kiae665. doi: 10.1093/plphys/kiae665 PMID: 39345498 Free PMC article. Updated. Preprint.
-
Mass spectrometry based proteomics for developmental neurobiology in the amphibian Xenopus laevis.Curr Top Dev Biol. 2021;145:205-231. doi: 10.1016/bs.ctdb.2021.04.002. Epub 2021 May 25. Curr Top Dev Biol. 2021. PMID: 34074530 Free PMC article. Review.
References
-
- Abbatiello SE, Schilling B, Mani DR, Zimmerman LJ, Hall SC, MacLean B, Albertolle M, Allen S, Burgess M, Cusack MP, Ghosh M, Hedrick V, Held JM, Inerowicz HD, Jackson A, Keshishian H, Kinsinger CR, Lyssand J, Makowski L, Mesri M, Rodriguez H, Rudnick P, Sadowski P, Sedransk N, Shaddox K, Skates SJ, Kuhn E, Smith D, Whiteaker JR, Whitwell C, Zhang S, Borchers CH, Fisher SJ, Gibson BW, Liebler DC, MacCoss MJ, Neubert TA, Paulovich AG, Regnier FE, Tempst P, Carr SA. Large-Scale Inter-Laboratory Study to Develop, Analytically Validate and Apply Highly Multiplexed, Quantitative Peptide Assays to Measure Cancer-Relevant Proteins in Plasma. Molecular & Cellular Proteomics. 2015;14(9):2357–74. - PMC - PubMed
-
- Abelin JG, Patel J, Lu X, Feeney CM, Fagbami L, Creech AL, Hu R, Lam D, Davison D, Pino L, Qiao JW, Kuhn E, Officer A, Li J, Abbatiello S, Subramanian A, Sidman R, Snyder E, Carr SA, Jaffe JD. Reduced-representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-scale Comparison of Drug-induced Phenotypes. Molecular & Cellular Proteomics. 2016;15(5):1622–41. - PMC - PubMed
-
- Anderson L, Hunter CL. Quantitative Mass Spectrometric Multiple Reaction Monitoring Assays for Major Plasma Proteins. Molecular & Cellular Proteomics. 2005;5(4):573–588. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
