

Host and viral traits predict zoonotic spillover from mammals
- PMID: 28636590
- PMCID: PMC5570460
- DOI: 10.1038/nature22975
Host and viral traits predict zoonotic spillover from mammals
Erratum in
-
Erratum: Host and viral traits predict zoonotic spillover from mammals.Nature. 2017 Aug 31;548(7669):612. doi: 10.1038/nature23660. Epub 2017 Aug 23. Nature. 2017. PMID: 29411779 Free PMC article.
Abstract
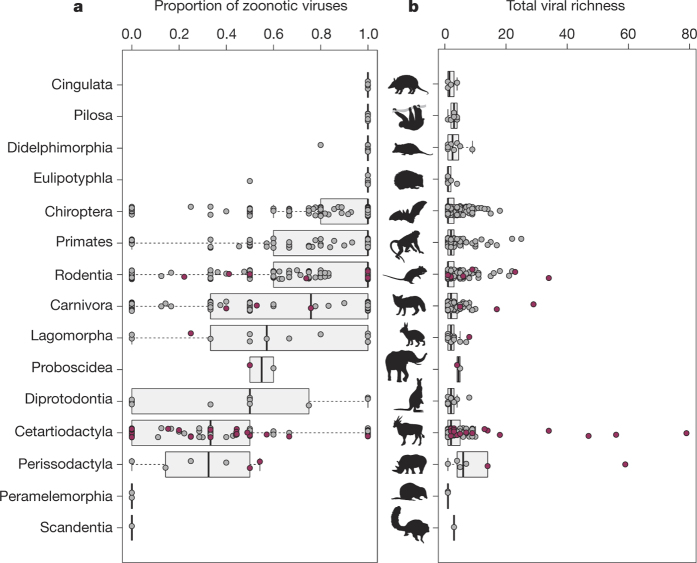
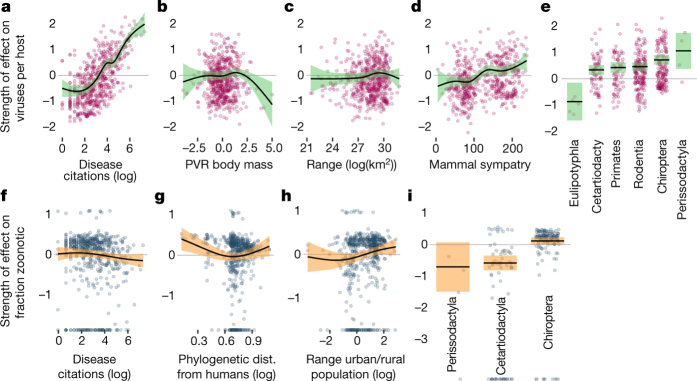
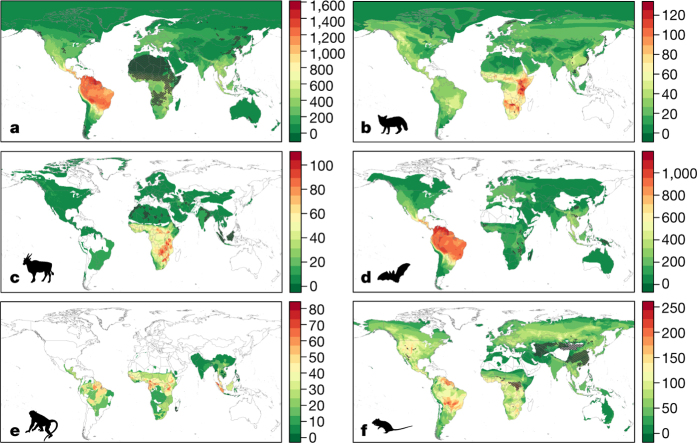
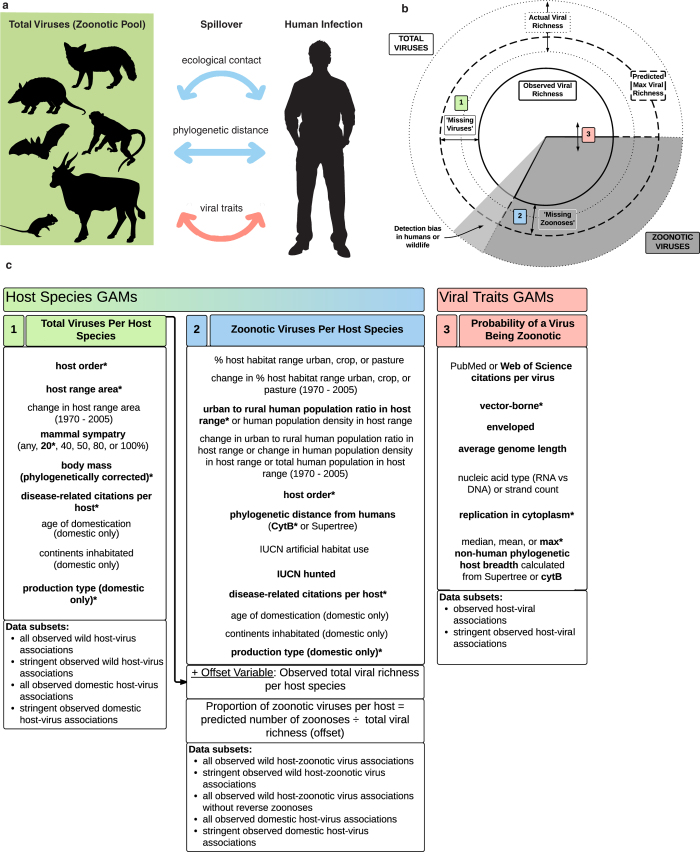
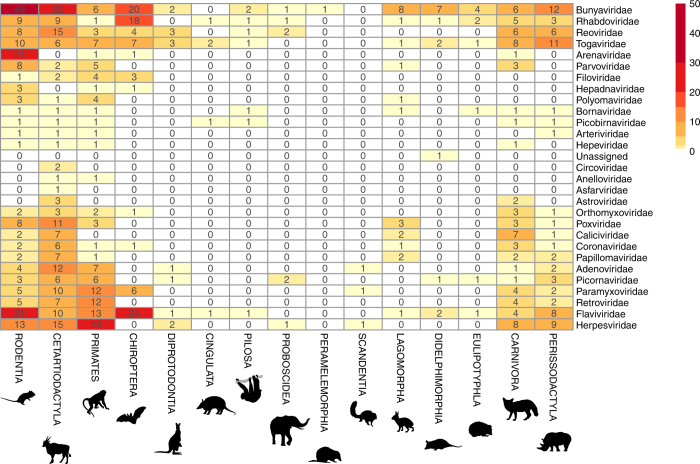
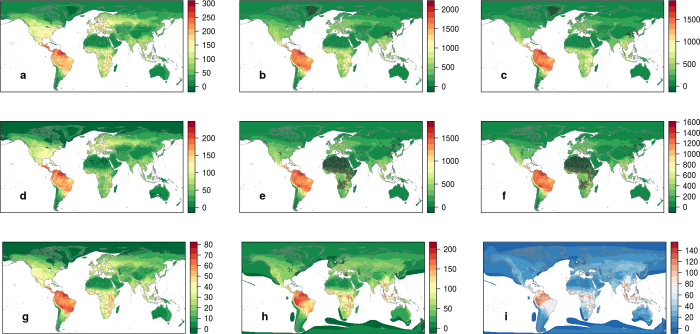
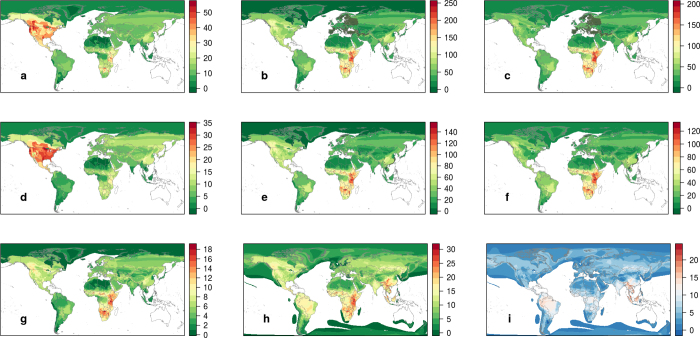
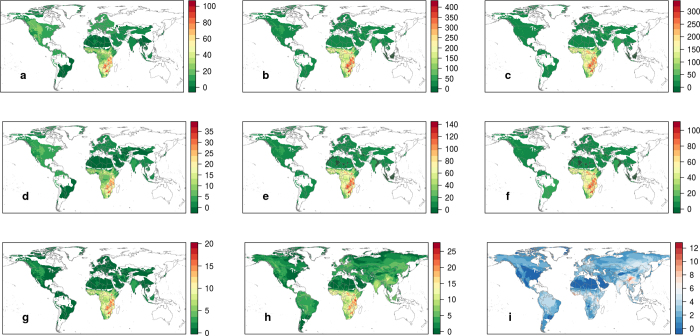
The majority of human emerging infectious diseases are zoonotic, with viruses that originate in wild mammals of particular concern (for example, HIV, Ebola and SARS). Understanding patterns of viral diversity in wildlife and determinants of successful cross-species transmission, or spillover, are therefore key goals for pandemic surveillance programs. However, few analytical tools exist to identify which host species are likely to harbour the next human virus, or which viruses can cross species boundaries. Here we conduct a comprehensive analysis of mammalian host-virus relationships and show that both the total number of viruses that infect a given species and the proportion likely to be zoonotic are predictable. After controlling for research effort, the proportion of zoonotic viruses per species is predicted by phylogenetic relatedness to humans, host taxonomy and human population within a species range-which may reflect human-wildlife contact. We demonstrate that bats harbour a significantly higher proportion of zoonotic viruses than all other mammalian orders. We also identify the taxa and geographic regions with the largest estimated number of 'missing viruses' and 'missing zoonoses' and therefore of highest value for future surveillance. We then show that phylogenetic host breadth and other viral traits are significant predictors of zoonotic potential, providing a novel framework to assess if a newly discovered mammalian virus could infect people.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

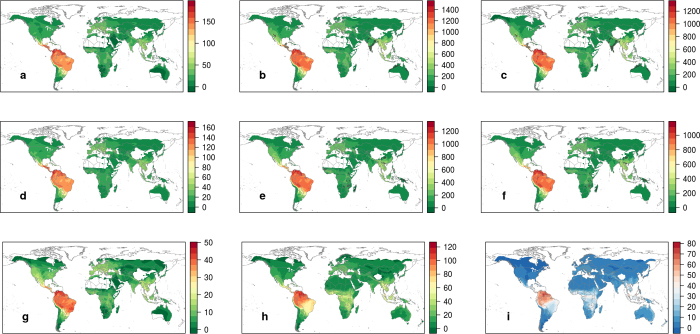

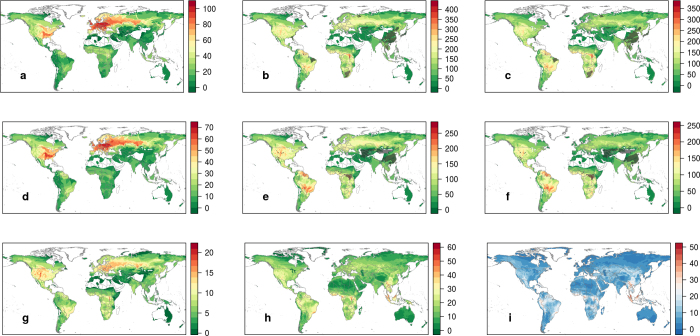

Comment in
-
Infectious diseases: Predictions of virus spillover across species.Nature. 2017 Jun 29;546(7660):603-604. doi: 10.1038/nature23088. Epub 2017 Jun 21. Nature. 2017. PMID: 28636591 No abstract available.
Similar articles
-
Spillover and pandemic properties of zoonotic viruses with high host plasticity.Sci Rep. 2015 Oct 7;5:14830. doi: 10.1038/srep14830. Sci Rep. 2015. PMID: 26445169 Free PMC article.
-
Mammals, wildlife trade, and the next global pandemic.Curr Biol. 2021 Aug 23;31(16):3671-3677.e3. doi: 10.1016/j.cub.2021.06.006. Epub 2021 Jul 7. Curr Biol. 2021. PMID: 34237267
-
A strategy to estimate unknown viral diversity in mammals.mBio. 2013 Sep 3;4(5):e00598-13. doi: 10.1128/mBio.00598-13. mBio. 2013. PMID: 24003179 Free PMC article.
-
Sialic Acid Receptors: The Key to Solving the Enigma of Zoonotic Virus Spillover.Viruses. 2021 Feb 8;13(2):262. doi: 10.3390/v13020262. Viruses. 2021. PMID: 33567791 Free PMC article. Review.
-
What do studies in wild mammals tell us about human emerging viral diseases in Mexico?Transbound Emerg Dis. 2020 Jan;67(1):33-45. doi: 10.1111/tbed.13336. Epub 2019 Oct 7. Transbound Emerg Dis. 2020. PMID: 31461573 Free PMC article. Review.
Cited by
-
Computer-Aided Prediction of the Interactions of Viral Proteases with Antiviral Drugs: Antiviral Potential of Broad-Spectrum Drugs.Molecules. 2023 Dec 31;29(1):225. doi: 10.3390/molecules29010225. Molecules. 2023. PMID: 38202808 Free PMC article.
-
A strategy to prevent future epidemics similar to the 2019-nCoV outbreak.Biosaf Health. 2020 Mar;2(1):6-8. doi: 10.1016/j.bsheal.2020.01.003. Epub 2020 Feb 5. Biosaf Health. 2020. PMID: 32562482 Free PMC article.
-
Addressing the illegal wildlife trade in the European Union as a public health issue to draw decision makers attention.Biol Conserv. 2020 Nov;251:108798. doi: 10.1016/j.biocon.2020.108798. Epub 2020 Oct 12. Biol Conserv. 2020. PMID: 33071292 Free PMC article. Review.
-
Research and Innovation Opportunities to Improve Epidemiological Knowledge and Control of Environmentally Driven Zoonoses.Ann Glob Health. 2022 Oct 21;88(1):93. doi: 10.5334/aogh.3770. eCollection 2022. Ann Glob Health. 2022. PMID: 36348706 Free PMC article. Review.
-
Venezuelan Equine Encephalitis Complex Alphavirus in Bats, French Guiana.Emerg Infect Dis. 2021 Apr;27(4):1141-5. doi: 10.3201/eid2704.202676. Emerg Infect Dis. 2021. PMID: 33756099 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
