

Newcastle Disease Virus Establishes Persistent Infection in Tumor Cells In Vitro: Contribution of the Cleavage Site of Fusion Protein and Second Sialic Acid Binding Site of Hemagglutinin-Neuraminidase
- PMID: 28592535
- PMCID: PMC5533931
- DOI: 10.1128/JVI.00770-17
Newcastle Disease Virus Establishes Persistent Infection in Tumor Cells In Vitro: Contribution of the Cleavage Site of Fusion Protein and Second Sialic Acid Binding Site of Hemagglutinin-Neuraminidase
Abstract
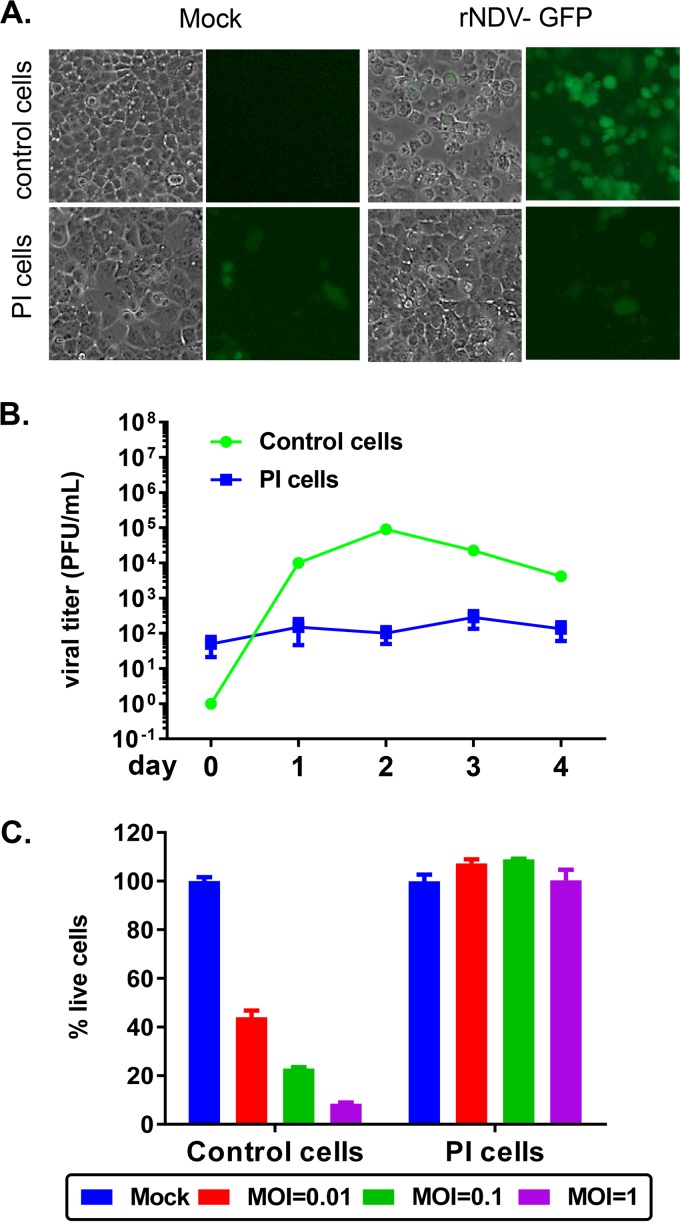
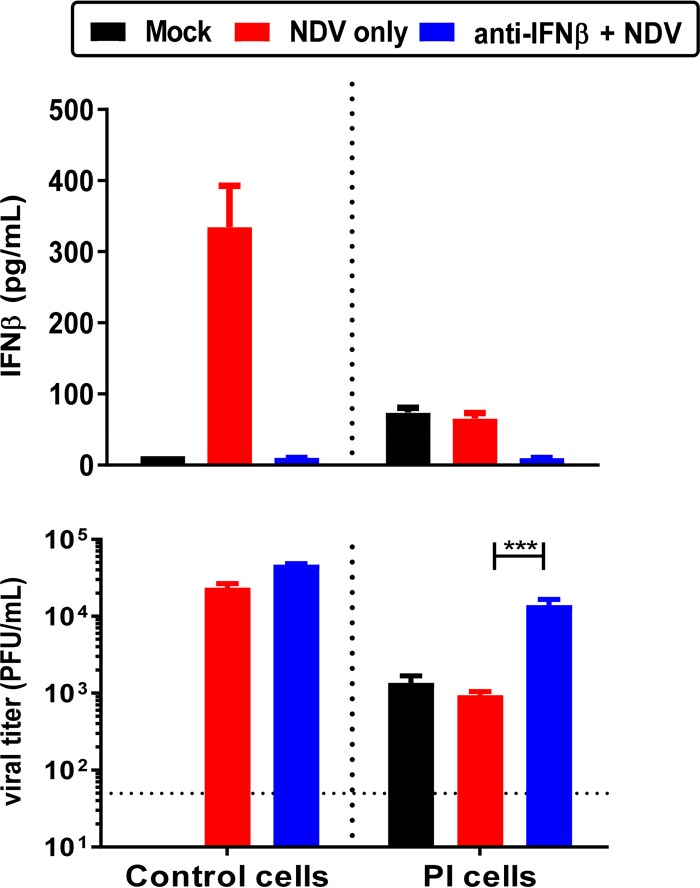
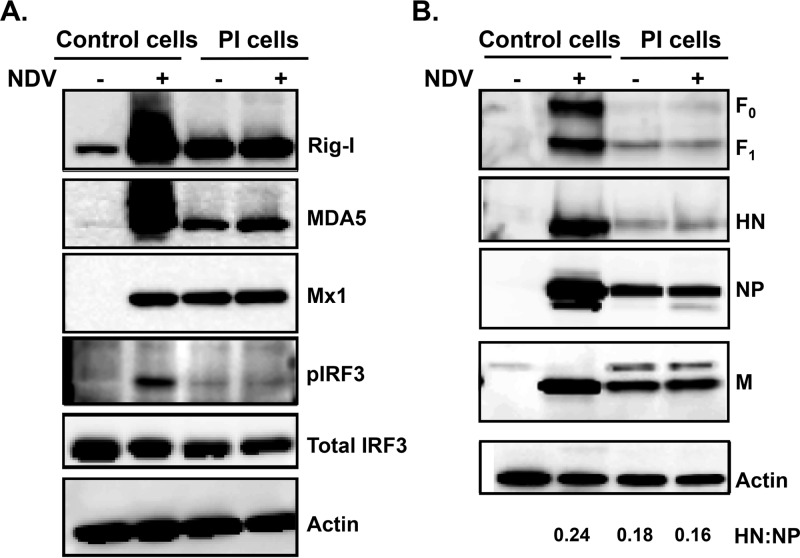
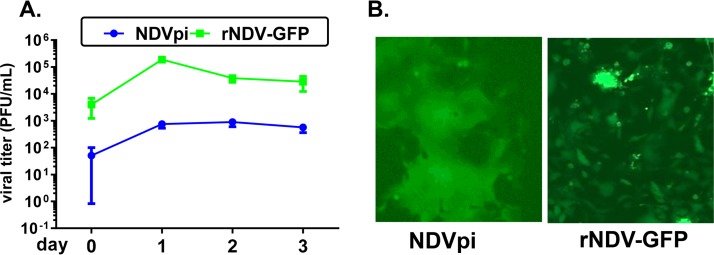
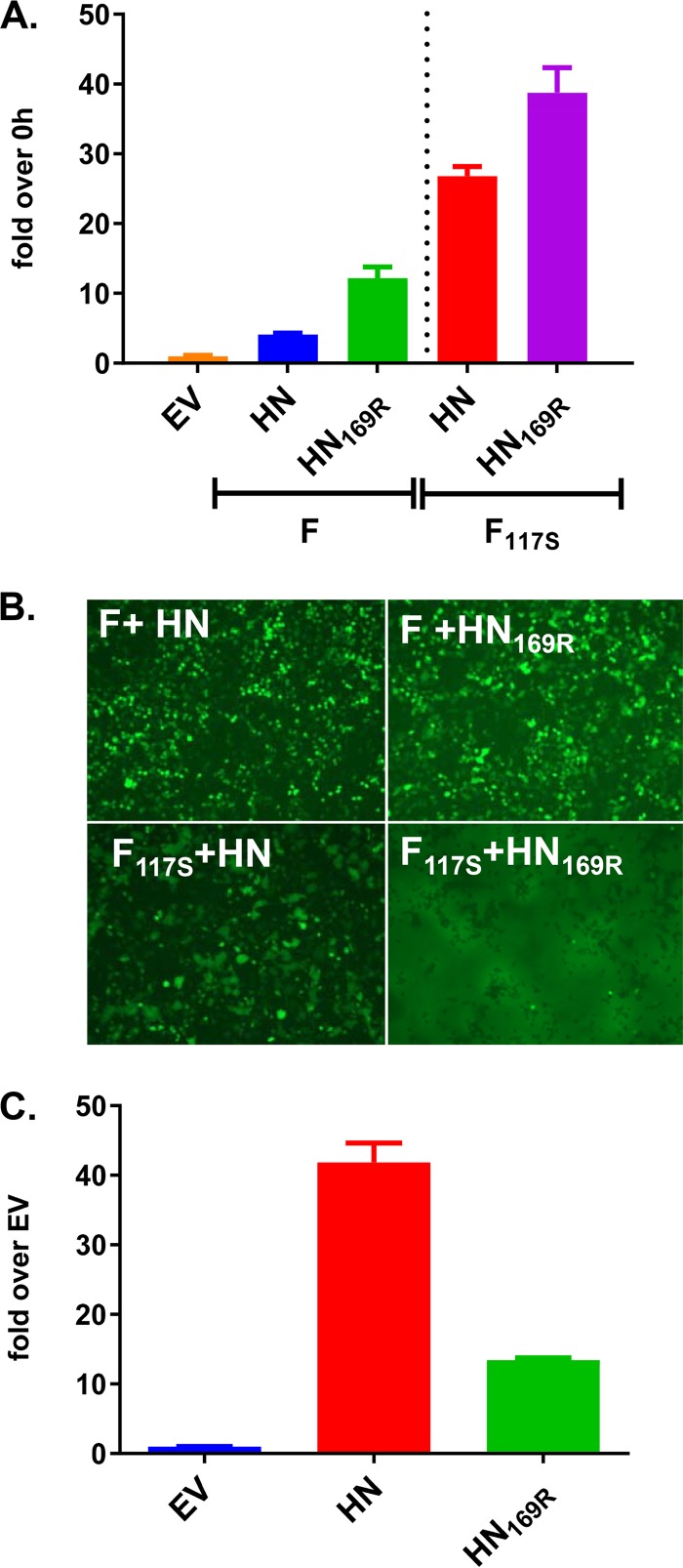
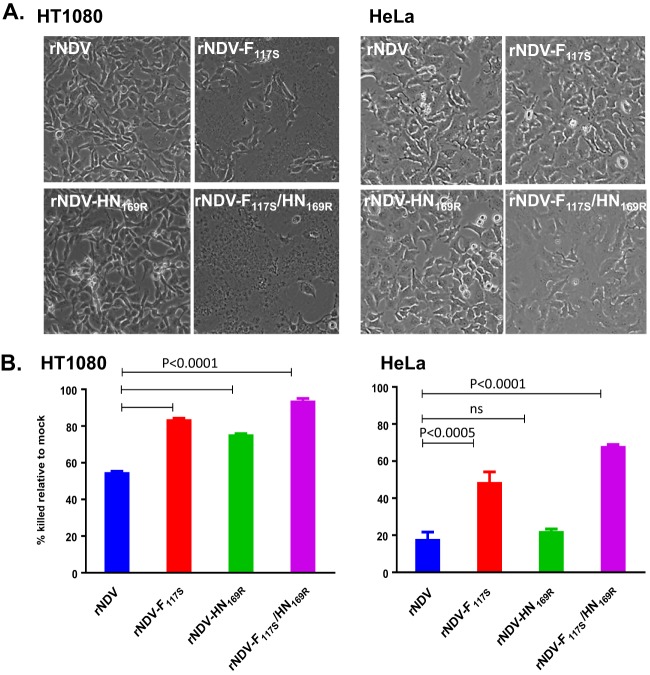
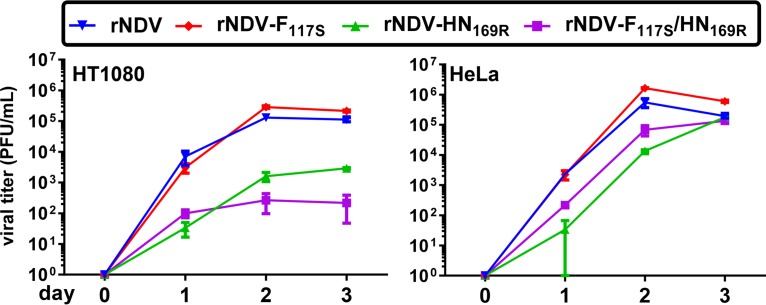
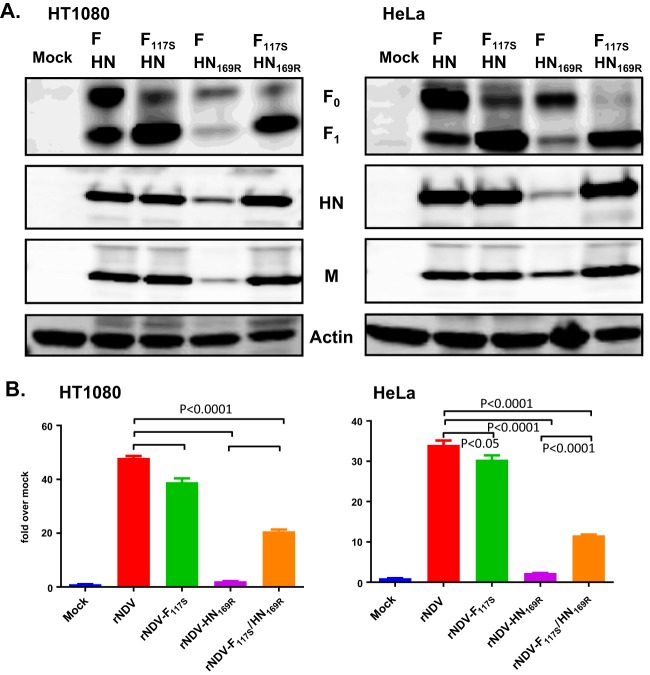
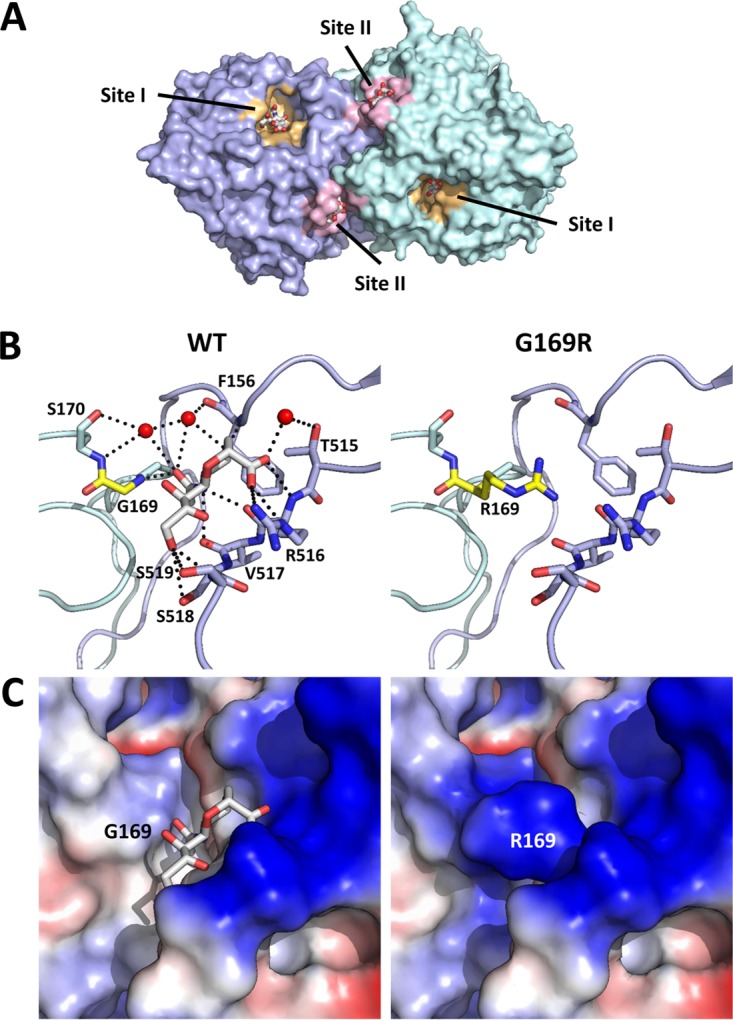
Newcastle disease virus (NDV) is an oncolytic virus being developed for the treatment of cancer. Following infection of a human ovarian cancer cell line (OVCAR3) with a recombinant low-pathogenic NDV, persistent infection was established in a subset of tumor cells. Persistently infected (PI) cells exhibited resistance to superinfection with NDV and established an antiviral state, as demonstrated by upregulation of interferon and interferon-induced genes such as myxoma resistance gene 1 (Mx1) and retinoic acid-inducing gene-I (RIG-I). Viruses released from PI cells induced higher cell-to-cell fusion than the parental virus following infection in two tumor cell lines tested, HT1080 and HeLa, and remained attenuated in chickens. Two mutations, one in the fusion (F) protein cleavage site, F117S (F117S), and another in hemagglutinin-neuraminidase (HN), G169R (HN169R), located in the second sialic acid binding region, were responsible for the hyperfusogenic phenotype. F117S improves F protein cleavage efficiency, facilitating cell-to-cell fusion, while HN169R possesses a multifaceted role in contributing to higher fusion, reduced receptor binding, and lower neuraminidase activity, which together result in increased fusion and reduced viral replication. Thus, establishment of persistent infection in vitro involves viral genetic changes that facilitate efficient viral spread from cell to cell as a potential mechanism to escape host antiviral responses. The results of our study also demonstrate a critical role in the viral life cycle for the second receptor binding region of the HN protein, which is conserved in several paramyxoviruses.IMPORTANCE Oncolytic Newcastle disease virus (NDV) could establish persistent infection in a tumor cell line, resulting in a steady antiviral state reflected by constitutively expressed interferon. Viruses isolated from persistently infected cells are highly fusogenic, and this phenotype has been mapped to two mutations, one each in the fusion (F) and hemagglutinin-neuraminidase (HN) proteins. The F117S mutation in the F protein cleavage site improved F protein cleavage efficiency while the HN169R mutation located at the second receptor binding site of the HN protein contributed to a complex phenotype consisting of a modest increase in fusion and cell killing, lower neuraminidase activity, and reduced viral growth. This study highlights the intricate nature of these two mutations in the glycoproteins of NDV in the establishment of persistent infection. The data also shed light on the critical balance between the F and HN proteins required for efficient NDV infection and their role in avian pathogenicity.
Keywords: NDV; Newcastle disease virus; fusion; paramyxovirus; persistent infection.
Copyright © 2017 American Society for Microbiology.
Figures
Similar articles
-
Roles of the highly conserved amino acids in the globular head and stalk region of the Newcastle disease virus HN protein in the membrane fusion process.Biosci Trends. 2015 Feb;9(1):56-64. doi: 10.5582/bst.2014.01140. Biosci Trends. 2015. PMID: 25787910
-
Roles of the fusion and hemagglutinin-neuraminidase proteins in replication, tropism, and pathogenicity of avian paramyxoviruses.J Virol. 2011 Sep;85(17):8582-96. doi: 10.1128/JVI.00652-11. Epub 2011 Jun 15. J Virol. 2011. PMID: 21680512 Free PMC article.
-
Hemagglutinin-Neuraminidase and fusion genes are determinants of NDV thermostability.Vet Microbiol. 2019 Jan;228:53-60. doi: 10.1016/j.vetmic.2018.11.013. Epub 2018 Nov 19. Vet Microbiol. 2019. PMID: 30593380
-
Structure and function of a paramyxovirus fusion protein.Biochim Biophys Acta. 2003 Jul 11;1614(1):73-84. doi: 10.1016/s0005-2736(03)00164-0. Biochim Biophys Acta. 2003. PMID: 12873767 Review.
-
Paramyxovirus membrane fusion: lessons from the F and HN atomic structures.Virology. 2006 Jan 5;344(1):30-7. doi: 10.1016/j.virol.2005.09.007. Virology. 2006. PMID: 16364733 Free PMC article. Review.
Cited by
-
Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy.Biomedicines. 2019 Aug 30;7(3):66. doi: 10.3390/biomedicines7030066. Biomedicines. 2019. PMID: 31480379 Free PMC article. Review.
-
Parainfluenza Virus Infection Sensitizes Cancer Cells to DNA-Damaging Agents: Implications for Oncolytic Virus Therapy.J Virol. 2018 Mar 14;92(7):e01948-17. doi: 10.1128/JVI.01948-17. Print 2018 Apr 1. J Virol. 2018. PMID: 29343567 Free PMC article.
-
Biological Significance of Dual Mutations A494D and E495K of the Genotype III Newcastle Disease Virus Hemagglutinin-Neuraminidase In Vitro and In Vivo.Viruses. 2022 Oct 25;14(11):2338. doi: 10.3390/v14112338. Viruses. 2022. PMID: 36366435 Free PMC article.
-
Syncytia Formation in Oncolytic Virotherapy.Mol Ther Oncolytics. 2019 Oct 1;15:131-139. doi: 10.1016/j.omto.2019.09.006. eCollection 2019 Dec 20. Mol Ther Oncolytics. 2019. PMID: 31890866 Free PMC article. Review.
-
Optimizing environmental safety and cell-killing potential of oncolytic Newcastle Disease virus with modifications of the V, F and HN genes.PLoS One. 2022 Feb 9;17(2):e0263707. doi: 10.1371/journal.pone.0263707. eCollection 2022. PLoS One. 2022. PMID: 35139115 Free PMC article.
References
-
- Stojdl DF, Lichty BD, ten Oever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275. doi:10.1016/S1535-6108(03)00241-1. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
