

Autophagy modulation: a potential therapeutic approach in cardiac hypertrophy
- PMID: 28576834
- PMCID: PMC5582915
- DOI: 10.1152/ajpheart.00145.2017
Autophagy modulation: a potential therapeutic approach in cardiac hypertrophy
Abstract
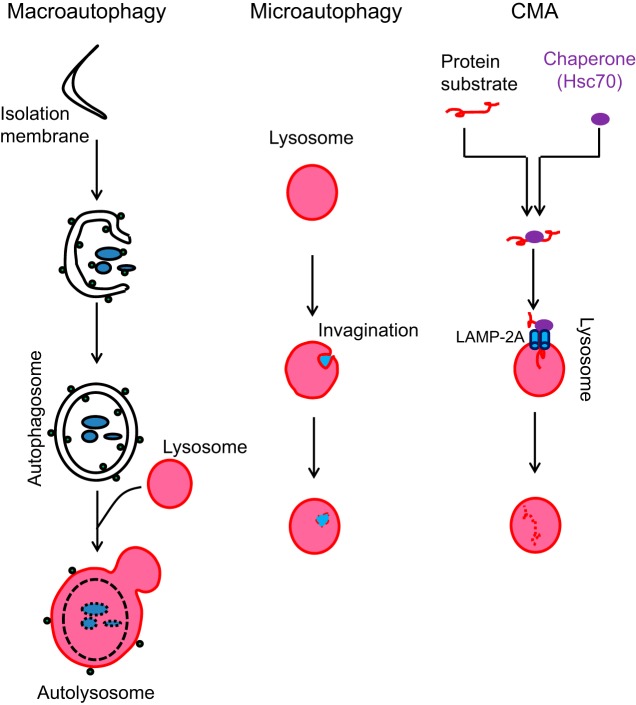
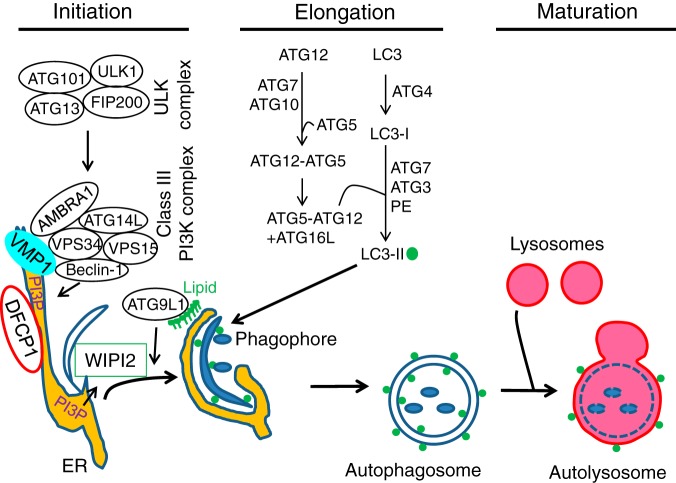
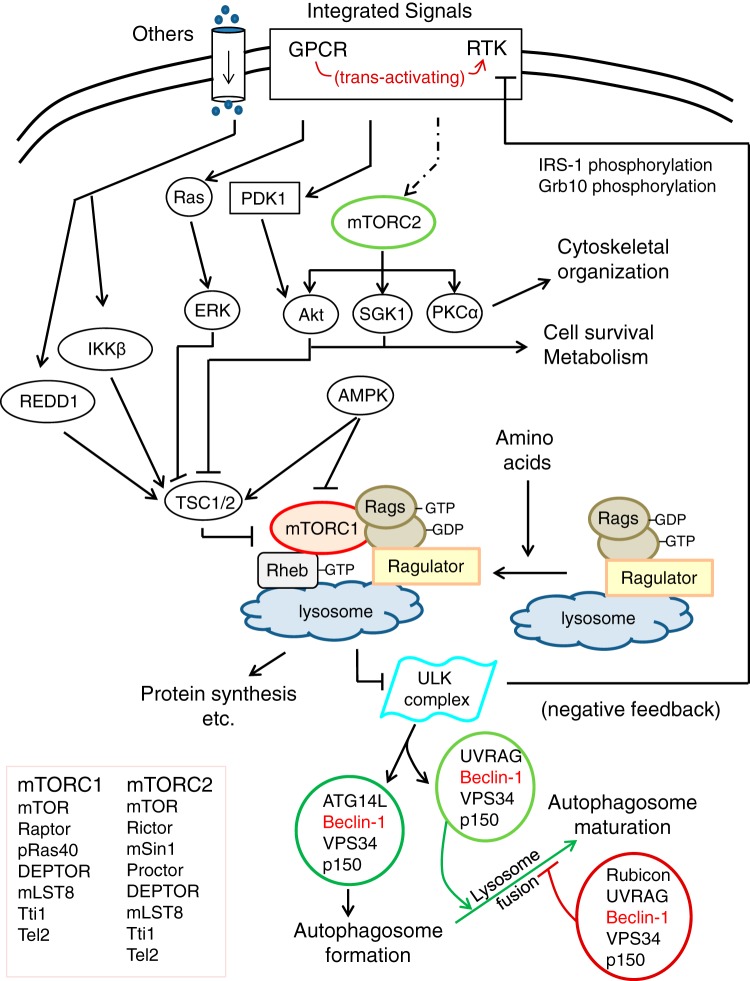
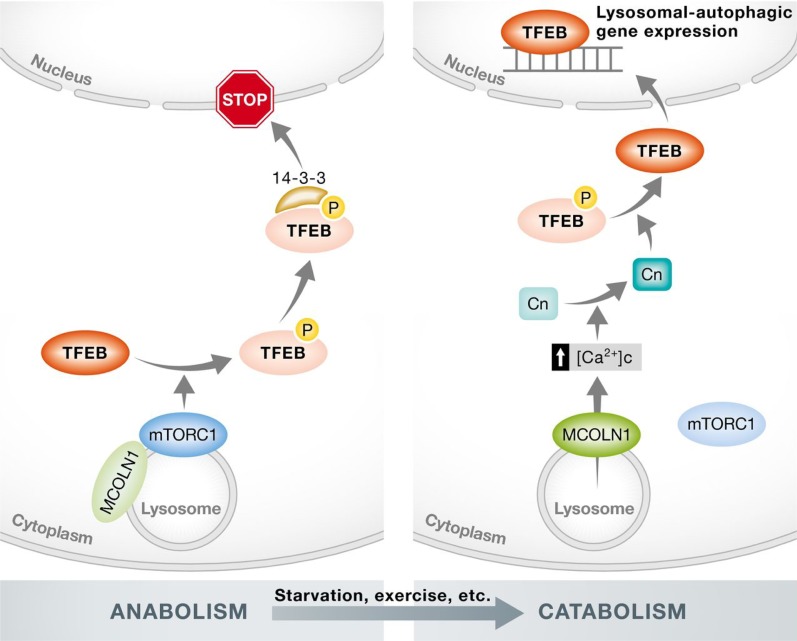
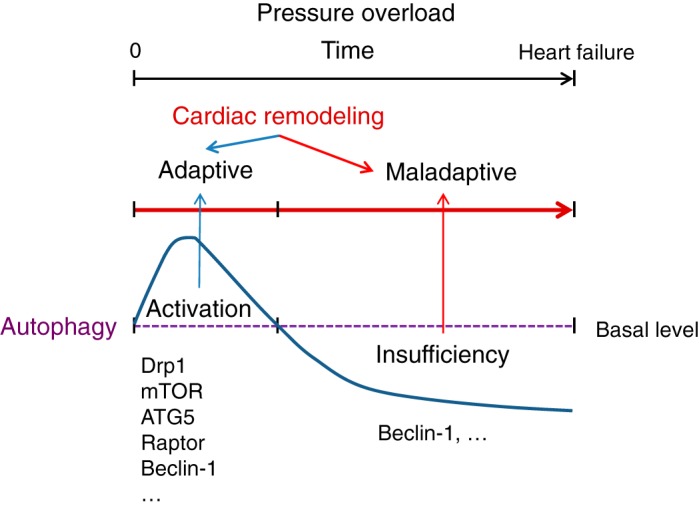
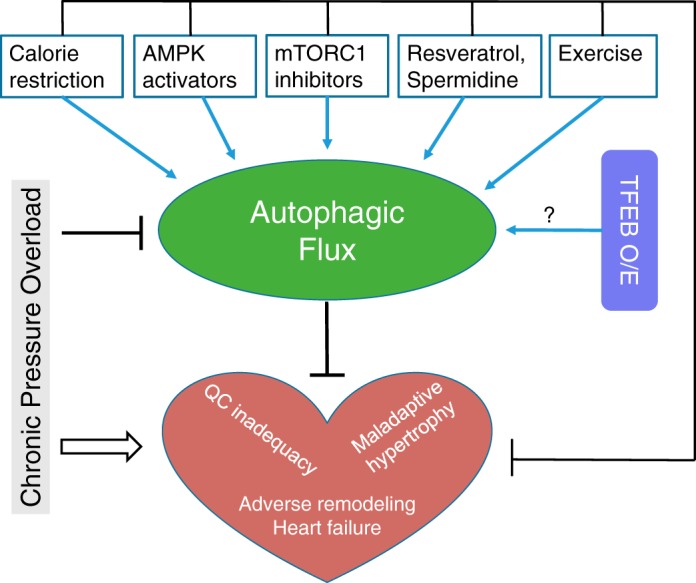
Autophagy is an evolutionarily conserved process used by the cell to degrade cytoplasmic contents for quality control, survival for temporal energy crisis, and catabolism and recycling. Rapidly increasing evidence has revealed an important pathogenic role of altered activity of the autophagosome-lysosome pathway (ALP) in cardiac hypertrophy and heart failure. Although an early study suggested that cardiac autophagy is increased and that this increase is maladaptive to the heart subject to pressure overload, more recent reports have overwhelmingly supported that myocardial ALP insufficiency results from chronic pressure overload and contributes to maladaptive cardiac remodeling and heart failure. This review examines multiple lines of preclinical evidence derived from recent studies regarding the role of autophagic dysfunction in pressure-overloaded hearts, attempts to reconcile the discrepancies, and proposes that resuming or improving ALP flux through coordinated enhancement of both the formation and the removal of autophagosomes would benefit the treatment of cardiac hypertrophy and heart failure resulting from chronic pressure overload.
Keywords: autophagy; cardiac hypertrophy; mechanistic target of rapamycin; pressure overload; transcription factor EB.
Copyright © 2017 the American Physiological Society.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the author(s).
Figures
Similar articles
-
Autophagy during cardiac remodeling.J Mol Cell Cardiol. 2016 Jun;95:11-8. doi: 10.1016/j.yjmcc.2015.12.003. Epub 2015 Dec 8. J Mol Cell Cardiol. 2016. PMID: 26678624 Review.
-
TFEB insufficiency promotes cardiac hypertrophy by blocking autophagic degradation of GATA4.J Biol Chem. 2021 Oct;297(4):101189. doi: 10.1016/j.jbc.2021.101189. Epub 2021 Sep 10. J Biol Chem. 2021. PMID: 34517007 Free PMC article.
-
Irisin alleviates pressure overload-induced cardiac hypertrophy by inducing protective autophagy via mTOR-independent activation of the AMPK-ULK1 pathway.J Mol Cell Cardiol. 2018 Aug;121:242-255. doi: 10.1016/j.yjmcc.2018.07.250. Epub 2018 Jul 24. J Mol Cell Cardiol. 2018. PMID: 30053525
-
Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy.Hypertension. 2014 Mar;63(3):490-9. doi: 10.1161/HYPERTENSIONAHA.113.02219. Epub 2013 Dec 23. Hypertension. 2014. PMID: 24366076 Free PMC article.
-
Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart.Cardiovasc Res. 2011 Feb 1;89(2):265-72. doi: 10.1093/cvr/cvq308. Epub 2010 Sep 28. Cardiovasc Res. 2011. PMID: 20880837 Review.
Cited by
-
Novel Insights into the Role of HDL-Associated Sphingosine-1-Phosphate in Cardiometabolic Diseases.Int J Mol Sci. 2019 Dec 12;20(24):6273. doi: 10.3390/ijms20246273. Int J Mol Sci. 2019. PMID: 31842389 Free PMC article. Review.
-
Tanshinone IIA Restores Dynamic Balance of Autophagosome/Autolysosome in Doxorubicin-Induced Cardiotoxicity via Targeting Beclin1/LAMP1.Cancers (Basel). 2019 Jun 28;11(7):910. doi: 10.3390/cancers11070910. Cancers (Basel). 2019. PMID: 31261758 Free PMC article.
-
Oridonin protects against cardiac hypertrophy by promoting P21-related autophagy.Cell Death Dis. 2019 May 24;10(6):403. doi: 10.1038/s41419-019-1617-y. Cell Death Dis. 2019. PMID: 31127082 Free PMC article.
-
Exacerbation of diabetic cardiac hypertrophy in OVE26 mice by angiotensin II is associated with JNK/c-Jun/miR-221-mediated autophagy inhibition.Oncotarget. 2017 Sep 28;8(63):106661-106671. doi: 10.18632/oncotarget.21302. eCollection 2017 Dec 5. Oncotarget. 2017. PMID: 29290979 Free PMC article.
-
Sacubitril/valsartan (LCZ696) ameliorates hyperthyroid-induced cardiac hypertrophy in male rats through modulation of miR-377, let-7 b, autophagy, and fibrotic signaling pathways.Sci Rep. 2022 Aug 27;12(1):14654. doi: 10.1038/s41598-022-18860-y. Sci Rep. 2022. PMID: 36030321 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical