

Inflammasomes and IL-1 biology in the pathogenesis of allograft dysfunction
- PMID: 28569730
- PMCID: PMC5451233
- DOI: 10.1172/JCI93537
Inflammasomes and IL-1 biology in the pathogenesis of allograft dysfunction
Abstract
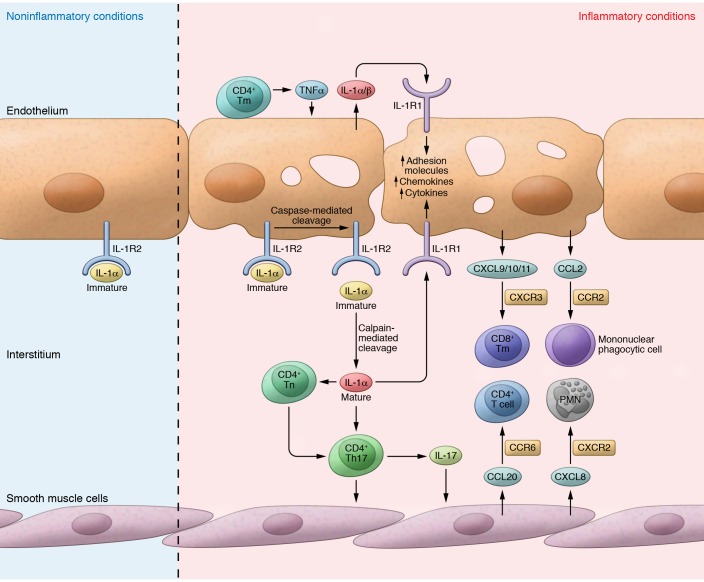
Inflammasomes are high-molecular-weight cytosolic complexes that mediate the activation of caspases. There are many inflammasomes, and each is influenced by a unique pattern-recognition receptor response. Two signals are typically involved in the inflammasome pathways. Signal one involves recognition of pathogen-associated molecular patterns (PAMPs), such as LPS or other colonizing/invading microbes, that interact with TLRs, which induce the downstream production of pro-IL-1β. This is followed by signal two, which involves recognition of PAMPs or damage-associated molecular patterns (DAMPs), such as uric acid or ATP, via NLRP3, which leads to caspase-1-dependent cleavage of pro-IL-1β to active IL-1β and pyroptosis. Ultimately, these two signals cause the release of multiple proinflammatory cytokines. Both PAMPs and DAMPs can be liberated by early insults to the allograft, including ischemia/reperfusion injury, infections, and rejection. The consequence of inflammasome activation and IL-1 expression is the upregulation of adhesion molecules and chemokines, which leads to allograft neutrophil sequestration, mononuclear phagocyte recruitment, and T cell activation, all of which are key steps in the continuum from allograft insult to chronic allograft dysfunction.
Conflict of interest statement
Figures

Similar articles
-
Inflammasome in Dendritic Cells Immunobiology: Implications to Diseases and Therapeutic Strategies.Curr Drug Targets. 2017;18(9):1003-1018. doi: 10.2174/1389450117666160921144830. Curr Drug Targets. 2017. PMID: 27660056 Review.
-
Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action.Microbiol Mol Biol Rev. 2018 Sep 12;82(4):e00015-18. doi: 10.1128/MMBR.00015-18. Print 2018 Dec. Microbiol Mol Biol Rev. 2018. PMID: 30209070 Free PMC article. Review.
-
Role of Inflammasome in Chronic Kidney Disease.Adv Exp Med Biol. 2019;1165:407-421. doi: 10.1007/978-981-13-8871-2_19. Adv Exp Med Biol. 2019. PMID: 31399976 Review.
-
Inflammasome as a promising therapeutic target for cancer.Life Sci. 2019 Aug 15;231:116593. doi: 10.1016/j.lfs.2019.116593. Epub 2019 Jun 19. Life Sci. 2019. PMID: 31228512 Review.
-
The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure.Brain Behav Immun. 2013 Feb;28:54-62. doi: 10.1016/j.bbi.2012.10.014. Epub 2012 Oct 24. Brain Behav Immun. 2013. PMID: 23103443
Cited by
-
Serum Uric Acid Levels and Risk of Eight Site-Specific Cancers: A Mendelian Randomization Study.Front Genet. 2021 Mar 9;12:608311. doi: 10.3389/fgene.2021.608311. eCollection 2021. Front Genet. 2021. PMID: 33767728 Free PMC article.
-
B cell-derived IL-1β and IL-6 drive T cell reconstitution following lymphoablation.Am J Transplant. 2020 Oct;20(10):2740-2754. doi: 10.1111/ajt.15960. Epub 2020 May 16. Am J Transplant. 2020. PMID: 32342598 Free PMC article.
-
Prevalence and significance of clonal hematopoiesis of indeterminate potential in lung transplant recipients.BMC Pulm Med. 2023 Oct 30;23(1):414. doi: 10.1186/s12890-023-02703-1. BMC Pulm Med. 2023. PMID: 37904125 Free PMC article.
-
Ex Vivo Lung Perfusion with β-Nicotinamide Adenine Dinucleotide (NAD+) Improves Ischemic Lung Function.Antioxidants (Basel). 2022 Apr 26;11(5):843. doi: 10.3390/antiox11050843. Antioxidants (Basel). 2022. PMID: 35624707 Free PMC article.
-
NLRP3 Inflammasome is Activated in Rat Pancreatic Islets by Transplantation and Hypoxia.Sci Rep. 2020 Apr 24;10(1):7011. doi: 10.1038/s41598-020-64054-9. Sci Rep. 2020. PMID: 32332867 Free PMC article.
References
-
- Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. 2006;83(2):447S–455S. - PubMed
