

Structural modeling of a novel SLC38A8 mutation that causes foveal hypoplasia
- PMID: 28546991
- PMCID: PMC5441399
- DOI: 10.1002/mgg3.266
Structural modeling of a novel SLC38A8 mutation that causes foveal hypoplasia
Abstract
Background: Foveal hypoplasia (FH) in the absence of albinism, aniridia, microphthalmia, or achromatopsia is exceedingly rare, and the molecular basis for the disorder remains unknown. FH is characterized by the absence of both the retinal foveal pit and avascular zone, but with preserved retinal architecture. SLC38A8 encodes a sodium-coupled neutral amino acid transporter with a preference for glutamate as a substrate. SLC38A8 has been linked to FH. Here, we describe a novel mutation to SLC38A8 which causes FH, and report the novel use of OCT-angiography to improve the precision of FH diagnosis. More so, we used computational modeling to explore possible functional effects of known SLC38A8 mutations.
Methods: Fundus autofluorescence, SD-OCT, and OCT-angiography were used to make the clinical diagnosis. Whole-exome sequencing led to the identification of a novel disease-causing variant in SLC38A8. Computational modeling approaches were used to visualize known SLC38A8 mutations, as well as to predict mutation effects on transporter structure and function.
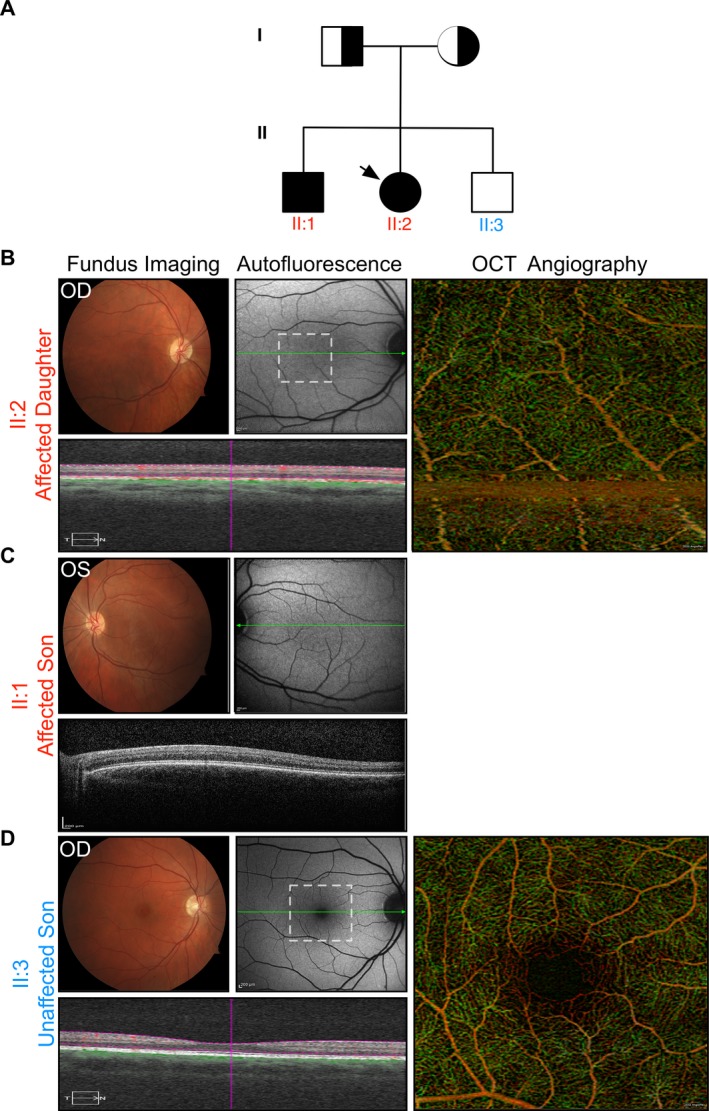
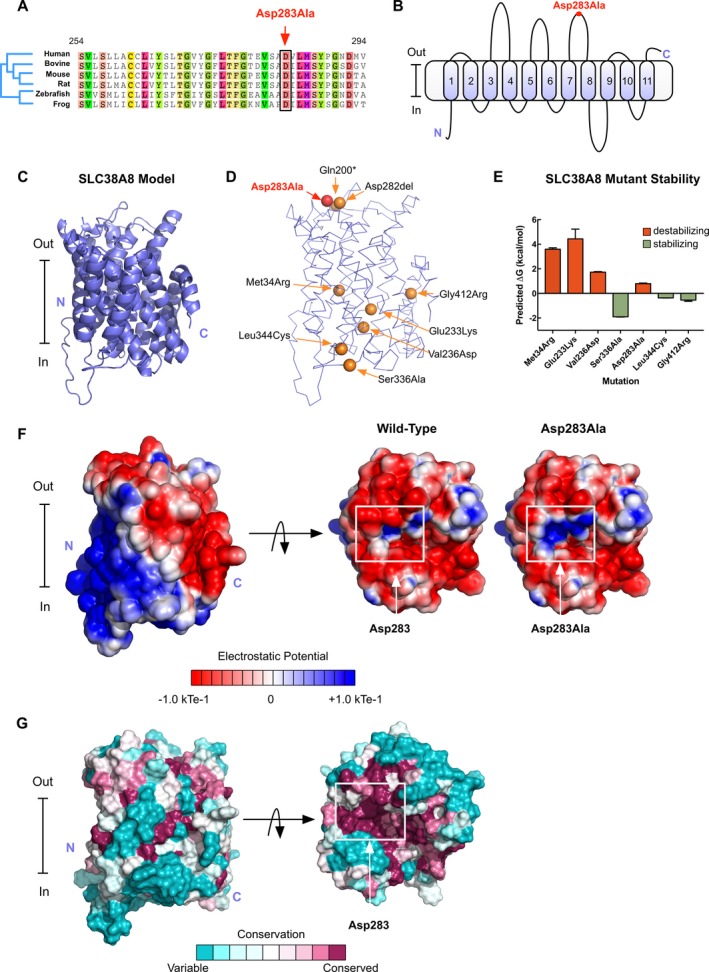
Results: We identified a novel point mutation in SLC38A8 that causes FH. A conclusive diagnosis was made using OCT-angiography, which more clearly revealed retinal vasculature penetrating into the foveal region. Structural modeling of the channel showed the mutation was near previously published mutations, clustered on an extracellular loop. Our modeling also predicted that the mutation destabilizes the protein by altering the electrostatic potential within the channel pore.
Conclusion: Our results demonstrate a novel use for OCT-angiography in confirming FH, and also uncover genotype-phenotype correlations of FH-linked SLC38A8 mutations.
Keywords: OCT‐angiography; SLC38A8; foveal hypoplasia; precision medicine; structural modeling.
Figures
Similar articles
-
Genotypic and Phenotypic Spectrum of Foveal Hypoplasia: A Multicenter Study.Ophthalmology. 2022 Jun;129(6):708-718. doi: 10.1016/j.ophtha.2022.02.010. Epub 2022 Feb 11. Ophthalmology. 2022. PMID: 35157951 Free PMC article.
-
Novel pathogenic variants of SLC38A8 gene and literature review.Eur J Ophthalmol. 2024 Nov;34(6):1740-1749. doi: 10.1177/11206721241242155. Epub 2024 Mar 22. Eur J Ophthalmol. 2024. PMID: 38515398 Review.
-
The pathogenicity of SLC38A8 in five families with foveal hypoplasia and congenital nystagmus.Exp Eye Res. 2020 Apr;193:107958. doi: 10.1016/j.exer.2020.107958. Epub 2020 Feb 4. Exp Eye Res. 2020. PMID: 32032626
-
Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism.Am J Hum Genet. 2013 Dec 5;93(6):1143-50. doi: 10.1016/j.ajhg.2013.11.002. Epub 2013 Nov 27. Am J Hum Genet. 2013. PMID: 24290379 Free PMC article. Review.
-
Foveal hypoplasia and optical coherence tomographic imaging.Taiwan J Ophthalmol. 2018 Oct-Dec;8(4):181-188. doi: 10.4103/tjo.tjo_101_18. Taiwan J Ophthalmol. 2018. PMID: 30637189 Free PMC article. Review.
Cited by
-
A Slc38a8 Mouse Model of FHONDA Syndrome Faithfully Recapitulates the Visual Deficits of Albinism Without Pigmentation Defects.Invest Ophthalmol Vis Sci. 2023 Oct 3;64(13):32. doi: 10.1167/iovs.64.13.32. Invest Ophthalmol Vis Sci. 2023. PMID: 37862028 Free PMC article.
-
Missense mutation in SLIT2 associated with congenital myopia, anisometropia, connective tissue abnormalities, and obesity.Orphanet J Rare Dis. 2018 Aug 15;13(1):138. doi: 10.1186/s13023-018-0885-4. Orphanet J Rare Dis. 2018. PMID: 30111362 Free PMC article.
-
SLC38A8 mutations result in arrested retinal development with loss of cone photoreceptor specialization.Hum Mol Genet. 2020 Nov 4;29(18):2989-3002. doi: 10.1093/hmg/ddaa166. Hum Mol Genet. 2020. PMID: 32744312 Free PMC article.
-
Genotype-Phenotype of Isolated Foveal Hypoplasia in a Large Cohort: Minor Iris Changes as an Indicator of PAX6 Involvement.Invest Ophthalmol Vis Sci. 2021 Aug 2;62(10):23. doi: 10.1167/iovs.62.10.23. Invest Ophthalmol Vis Sci. 2021. PMID: 34415986 Free PMC article.
-
Gene Therapy Restores Mfrp and Corrects Axial Eye Length.Sci Rep. 2017 Nov 23;7(1):16151. doi: 10.1038/s41598-017-16275-8. Sci Rep. 2017. PMID: 29170418 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
