

Conformational changes at cytoplasmic intersubunit interactions control Kir channel gating
- PMID: 28446610
- PMCID: PMC5473215
- DOI: 10.1074/jbc.M117.785154
Conformational changes at cytoplasmic intersubunit interactions control Kir channel gating
Abstract
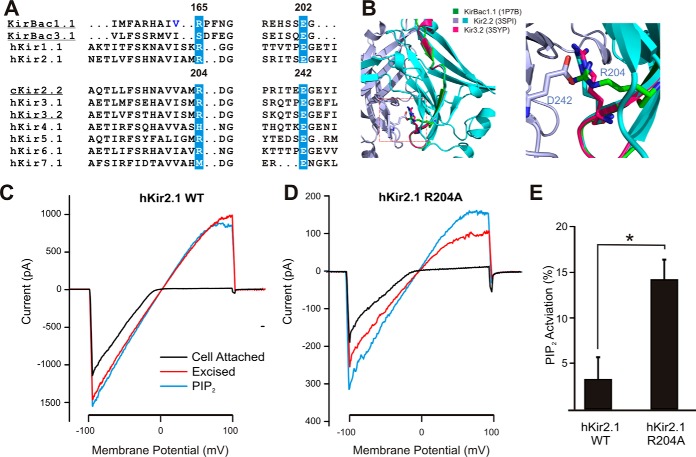
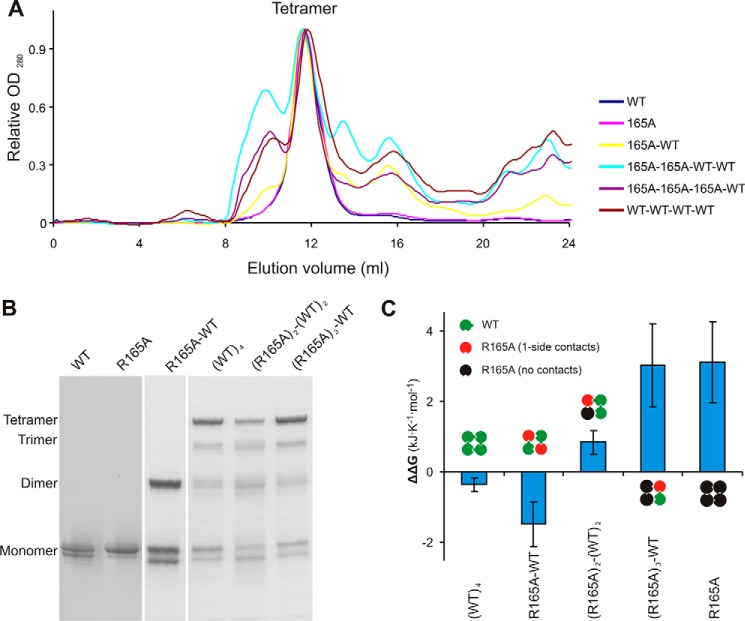
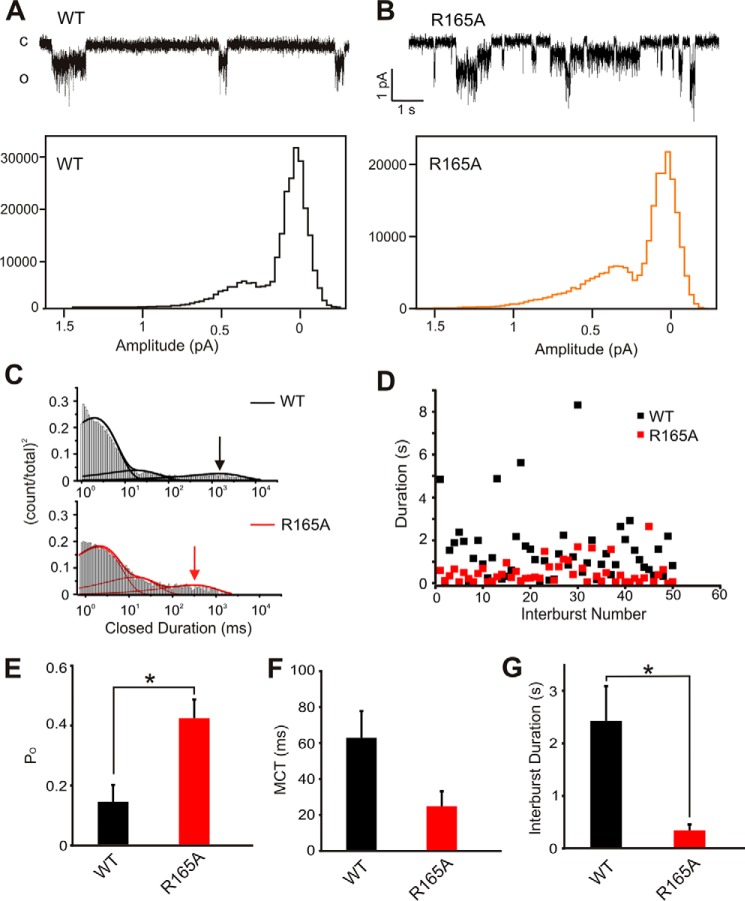
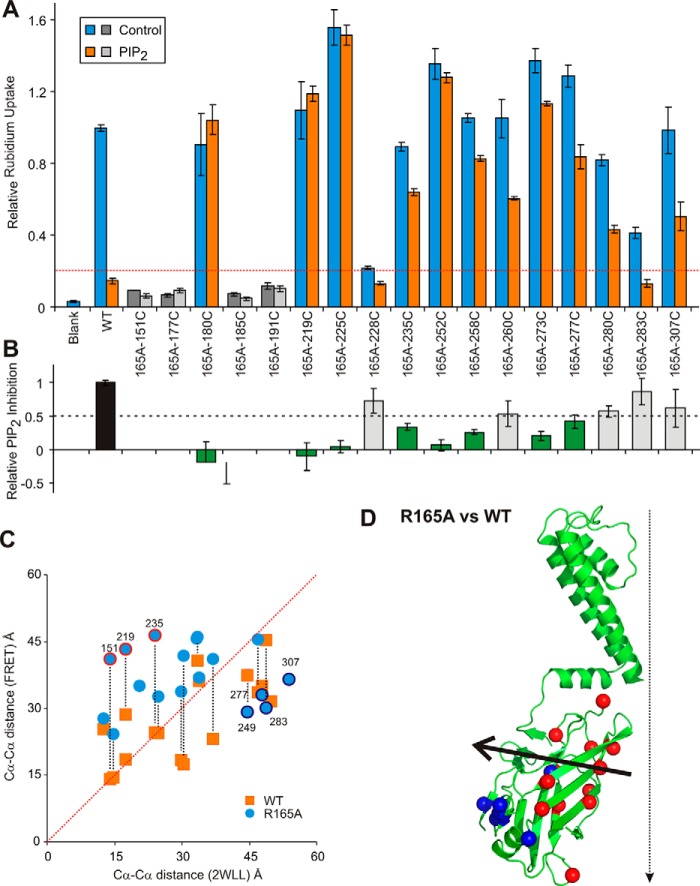
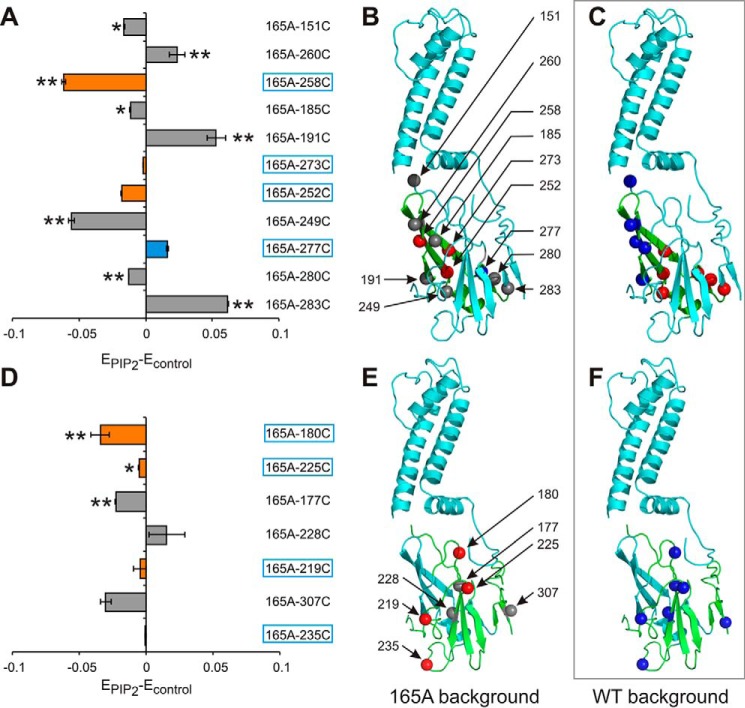
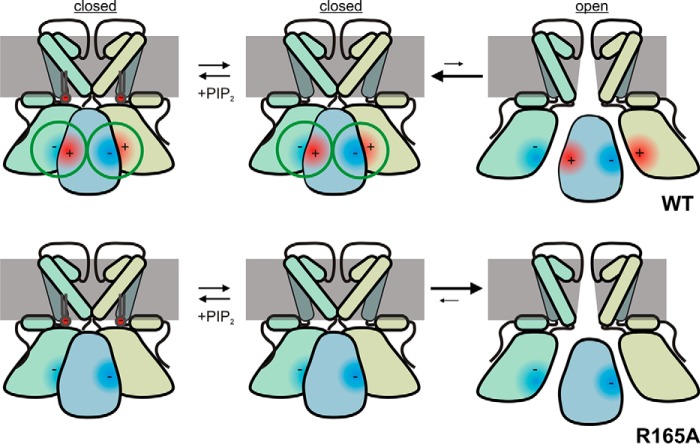
The defining structural feature of inward-rectifier potassium (Kir) channels is the unique Kir cytoplasmic domain. Recently we showed that salt bridges located at the cytoplasmic domain subunit interfaces (CD-Is) of eukaryotic Kir channels control channel gating via stability of a novel inactivated closed state. The cytoplasmic domains of prokaryotic and eukaryotic Kir channels show similar conformational rearrangements to the common gating ligand, phosphatidylinositol bisphosphate (PIP2), although these exhibit opposite coupling to opening and closing transitions. In Kir2.1, mutation of one of these CD-I salt bridge residues (R204A) reduces apparent PIP2 sensitivity of channel activity, and here we show that Ala or Cys substitutions of the functionally equivalent residue (Arg-165) in the prokaryotic Kir channel KirBac1.1 also significantly decrease sensitivity of the channel to PIP2 (by 5-30-fold). To further understand the structural basis of CD-I control of Kir channel gating, we examined the effect of the R165A mutation on PIP2-induced changes in channel function and conformation. Single-channel analyses indicated that the R165A mutation disrupts the characteristic long interburst closed state of reconstituted KirBac1.1 in giant liposomes, resulting in a higher open probability due to more frequent opening bursts. Intramolecular FRET measurements indicate that, relative to wild-type channels, the R165A mutation results in splaying of the cytoplasmic domains away from the central axis and that PIP2 essentially induces opposite motions of the major β-sheet in this channel mutant. We conclude that the removal of stabilizing CD-I salt bridges results in a collapsed state of the Kir domain.
Keywords: fluorescence resonance energy transfer (FRET); gating; membrane transporter reconstitution; phosphatidylinositol; potassium channel.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures

Similar articles
-
The unique structural characteristics of the Kir 7.1 inward rectifier potassium channel: a novel player in energy homeostasis control.Am J Physiol Cell Physiol. 2023 Mar 1;324(3):C694-C706. doi: 10.1152/ajpcell.00335.2022. Epub 2023 Jan 30. Am J Physiol Cell Physiol. 2023. PMID: 36717105 Free PMC article. Review.
-
Hydrogen sulfide inhibits Kir2 and Kir3 channels by decreasing sensitivity to the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2).J Biol Chem. 2018 Mar 9;293(10):3546-3561. doi: 10.1074/jbc.RA117.001679. Epub 2018 Jan 9. J Biol Chem. 2018. PMID: 29317494 Free PMC article.
-
Control of Kir channel gating by cytoplasmic domain interface interactions.J Gen Physiol. 2017 May 1;149(5):561-576. doi: 10.1085/jgp.201611719. Epub 2017 Apr 7. J Gen Physiol. 2017. PMID: 28389584 Free PMC article.
-
Conformational changes upon gating of KirBac1.1 into an open-activated state revealed by solid-state NMR and functional assays.Proc Natl Acad Sci U S A. 2020 Feb 11;117(6):2938-2947. doi: 10.1073/pnas.1915010117. Epub 2020 Jan 24. Proc Natl Acad Sci U S A. 2020. PMID: 31980523 Free PMC article.
-
Lipid agonism: The PIP2 paradigm of ligand-gated ion channels.Biochim Biophys Acta. 2015 May;1851(5):620-8. doi: 10.1016/j.bbalip.2015.01.011. Epub 2015 Jan 26. Biochim Biophys Acta. 2015. PMID: 25633344 Free PMC article. Review.
Cited by
-
Cloning, functional expression, and pharmacological characterization of inwardly rectifying potassium channels (Kir) from Apis mellifera.Sci Rep. 2024 Apr 3;14(1):7834. doi: 10.1038/s41598-024-58234-0. Sci Rep. 2024. PMID: 38570597 Free PMC article.
-
Unexpected Gating Behaviour of an Engineered Potassium Channel Kir.Front Mol Biosci. 2021 Jun 10;8:691901. doi: 10.3389/fmolb.2021.691901. eCollection 2021. Front Mol Biosci. 2021. PMID: 34179097 Free PMC article.
-
G protein βγ subunits play a critical role in the actions of amphetamine.Transl Psychiatry. 2019 Feb 11;9(1):81. doi: 10.1038/s41398-019-0387-8. Transl Psychiatry. 2019. PMID: 30745563 Free PMC article.
-
The unique structural characteristics of the Kir 7.1 inward rectifier potassium channel: a novel player in energy homeostasis control.Am J Physiol Cell Physiol. 2023 Mar 1;324(3):C694-C706. doi: 10.1152/ajpcell.00335.2022. Epub 2023 Jan 30. Am J Physiol Cell Physiol. 2023. PMID: 36717105 Free PMC article. Review.
References
-
- Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., and Kurachi Y. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 - PubMed
-
- Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476 - PubMed
-
- Tristani-Firouzi M., and Etheridge S. P. (2010) Kir 2.1 channelopathies: the Andersen-Tawil syndrome. Pflugers Arch. 460, 289–294 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
