

Expansions of Cytotoxic CD4+CD28- T Cells Drive Excess Cardiovascular Mortality in Rheumatoid Arthritis and Other Chronic Inflammatory Conditions and Are Triggered by CMV Infection
- PMID: 28303136
- PMCID: PMC5332470
- DOI: 10.3389/fimmu.2017.00195
Expansions of Cytotoxic CD4+CD28- T Cells Drive Excess Cardiovascular Mortality in Rheumatoid Arthritis and Other Chronic Inflammatory Conditions and Are Triggered by CMV Infection
Abstract
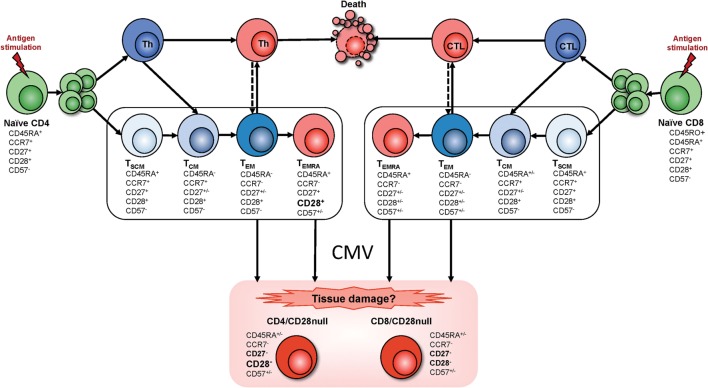
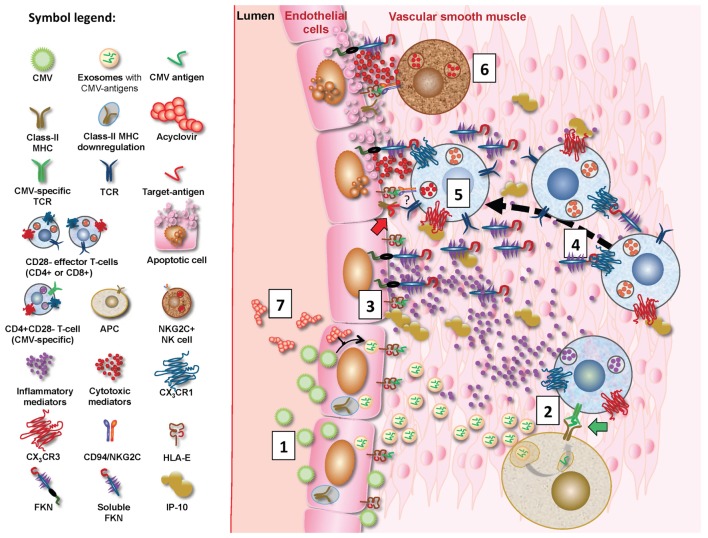
A large proportion of cardiovascular (CV) pathology results from immune-mediated damage, including systemic inflammation and cellular proliferation, which cause a narrowing of the blood vessels. Expansions of cytotoxic CD4+ T cells characterized by loss of CD28 ("CD4+CD28- T cells" or "CD4+CD28null cells") are closely associated with cardiovascular disease (CVD), in particular coronary artery damage. Direct involvement of these cells in damaging the vasculature has been demonstrated repeatedly. Moreover, CD4+CD28- T cells are significantly increased in rheumatoid arthritis (RA) and other autoimmune conditions. It is striking that expansions of this subset beyond 1-2% occur exclusively in CMV-infected people. CMV infection itself is known to increase the severity of autoimmune diseases, in particular RA and has also been linked to increased vascular pathology. A review of the recent literature on immunological changes in CVD, RA, and CMV infection provides strong evidence that expansions of cytotoxic CD4+CD28- T cells in RA and other chronic inflammatory conditions are limited to CMV-infected patients and driven by CMV infection. They are likely to be responsible for the excess CV mortality observed in these situations. The CD4+CD28- phenotype convincingly links CMV infection to CV mortality based on a direct cellular-pathological mechanism rather than epidemiological association.
Keywords: CD4 T cells; autoimmune diseases; cardiovascular diseases; chronic inflammatory disease; cytotoxic T cells.
Figures
Similar articles
-
CD28null pro-atherogenic CD4 T-cells explain the link between CMV infection and an increased risk of cardiovascular death.Theranostics. 2018 Aug 7;8(16):4509-4519. doi: 10.7150/thno.27428. eCollection 2018. Theranostics. 2018. PMID: 30214635 Free PMC article.
-
CD28 null CD4 T-cell expansions in autoimmune disease suggest a link with cytomegalovirus infection.F1000Res. 2019 Mar 25;8:F1000 Faculty Rev-327. doi: 10.12688/f1000research.17119.1. eCollection 2019. F1000Res. 2019. PMID: 30984377 Free PMC article. Review.
-
The host cellular immune response to cytomegalovirus targets the endothelium and is associated with increased arterial stiffness in ANCA-associated vasculitis.Arthritis Res Ther. 2018 Aug 29;20(1):194. doi: 10.1186/s13075-018-1695-8. Arthritis Res Ther. 2018. PMID: 30157919 Free PMC article.
-
Association of anticytomegalovirus seropositivity with more severe joint destruction and more frequent joint surgery in rheumatoid arthritis.Arthritis Rheum. 2012 Jun;64(6):1740-9. doi: 10.1002/art.34346. Epub 2011 Dec 19. Arthritis Rheum. 2012. PMID: 22183424
-
The immune system in extreme longevity.Exp Gerontol. 2008 Feb;43(2):61-5. doi: 10.1016/j.exger.2007.06.008. Epub 2007 Jul 4. Exp Gerontol. 2008. PMID: 17870272 Review.
Cited by
-
Abdominal Obesity-Related Disturbance of Insulin Sensitivity Is Associated with CD8+ EMRA Cells in the Elderly.Cells. 2021 Apr 23;10(5):998. doi: 10.3390/cells10050998. Cells. 2021. PMID: 33922813 Free PMC article.
-
Cytomegalovirus infection is associated with an increase in aortic stiffness in older men which may be mediated in part by CD4 memory T-cells.Theranostics. 2021 Mar 31;11(12):5728-5741. doi: 10.7150/thno.58356. eCollection 2021. Theranostics. 2021. PMID: 33897878 Free PMC article.
-
Social stressors associated with age-related T lymphocyte percentages in older US adults: Evidence from the US Health and Retirement Study.Proc Natl Acad Sci U S A. 2022 Jun 21;119(25):e2202780119. doi: 10.1073/pnas.2202780119. Epub 2022 Jun 13. Proc Natl Acad Sci U S A. 2022. PMID: 35696572 Free PMC article.
-
Coordinated Priming of NKG2D Pathway by IL-15 Enhanced Functional Properties of Cytotoxic CD4+CD28- T Cells Expanded in Systemic Lupus Erythematosus.Inflammation. 2023 Oct;46(5):1587-1601. doi: 10.1007/s10753-023-01860-z. Epub 2023 Jul 6. Inflammation. 2023. PMID: 37415045 Free PMC article.
-
Cytomegalovirus (CMV) Epitope-Specific CD4+ T Cells Are Inflated in HIV+ CMV+ Subjects.J Immunol. 2017 Nov 1;199(9):3187-3201. doi: 10.4049/jimmunol.1700851. Epub 2017 Oct 2. J Immunol. 2017. PMID: 28972094 Free PMC article.
References
-
- Vallejo AN, Brandes JC, Weyand CM, Goronzy JJ. Modulation of CD28 expression: distinct regulatory pathways during activation and replicative senescence. J Immunol (1999) 162(11):6572–9. - PubMed
-
- Liuzzo G, Kopecky SL, Frye RL, O’Fallon WM, Maseri A, Goronzy JJ, et al. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation (1999) 100(21):2135–9. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials