

Accurate model annotation of a near-atomic resolution cryo-EM map
- PMID: 28270620
- PMCID: PMC5373346
- DOI: 10.1073/pnas.1621152114
Accurate model annotation of a near-atomic resolution cryo-EM map
Abstract
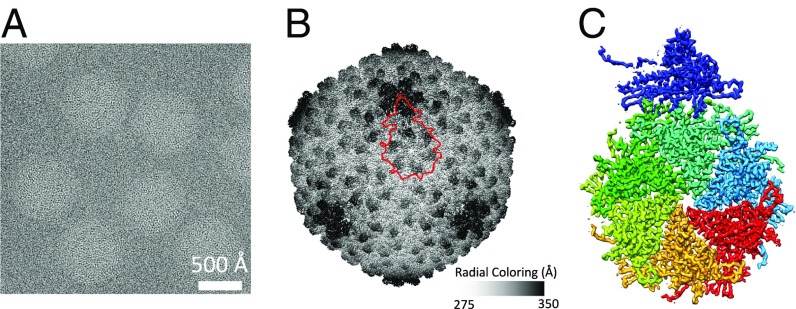
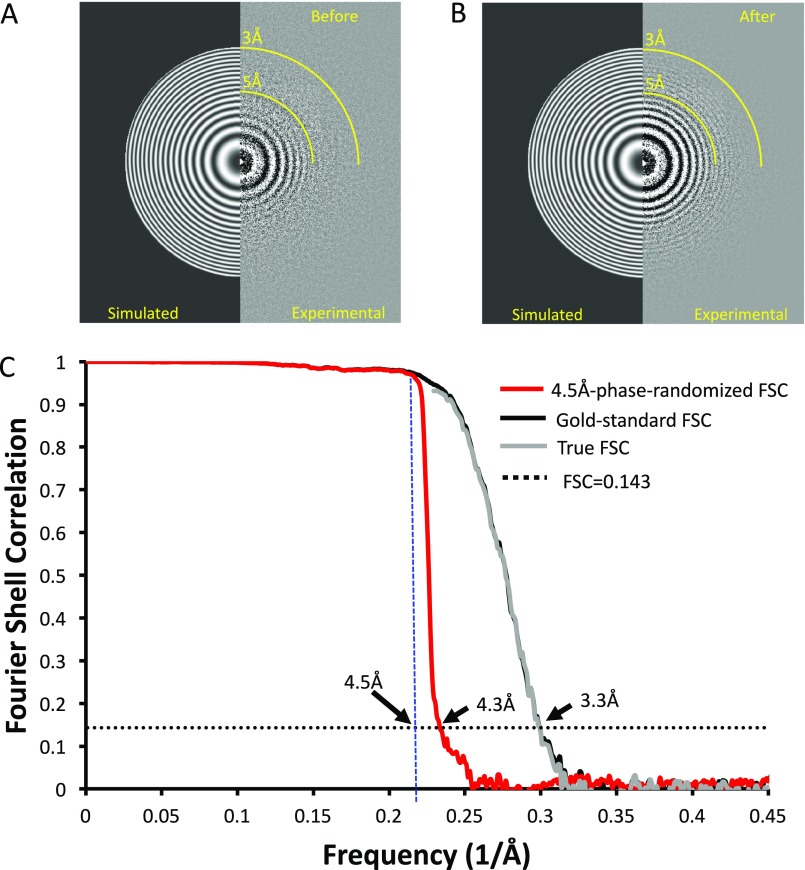
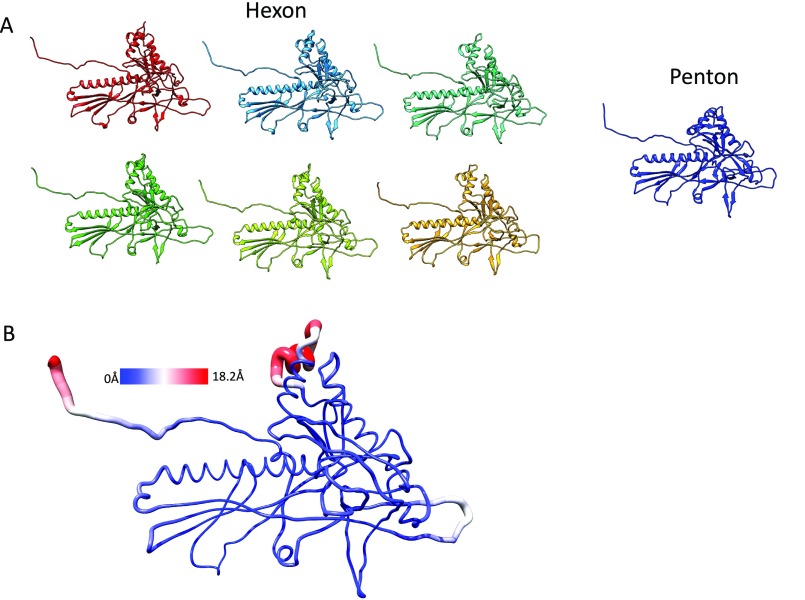
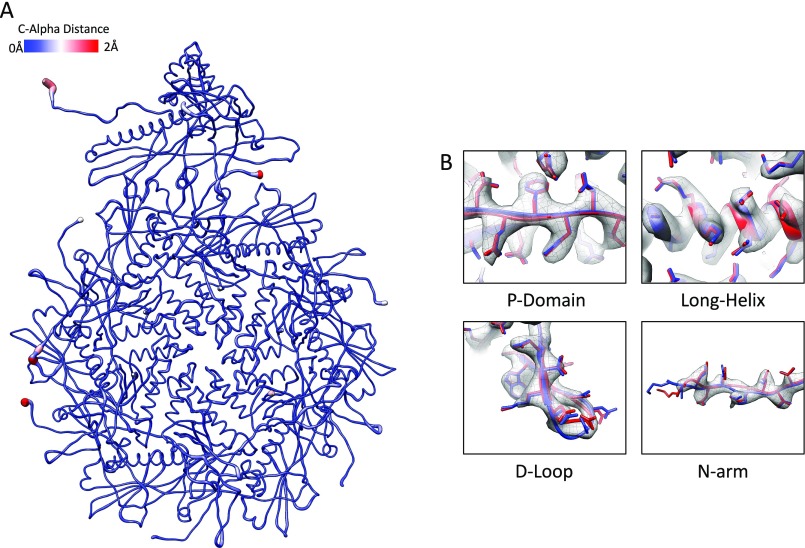
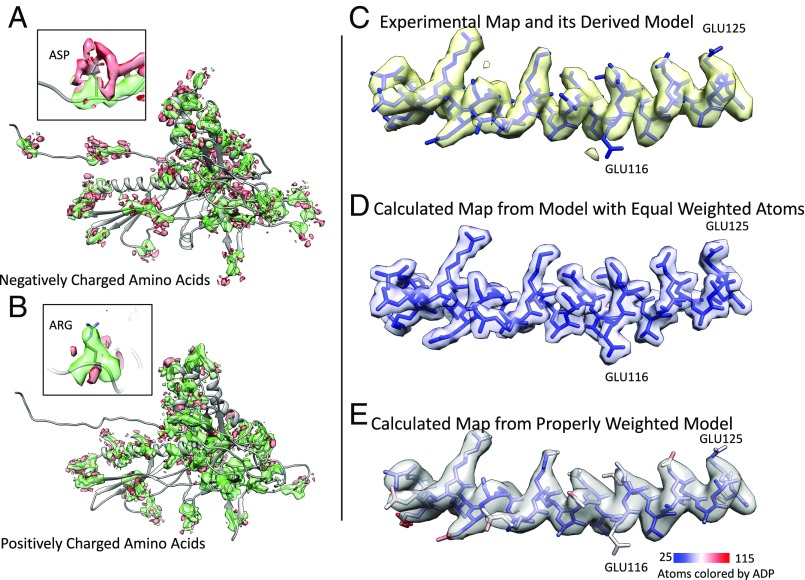
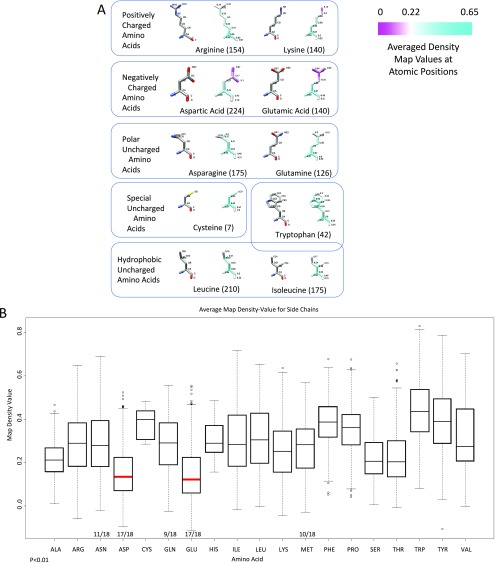
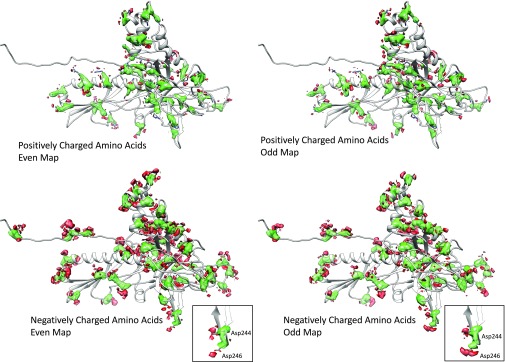
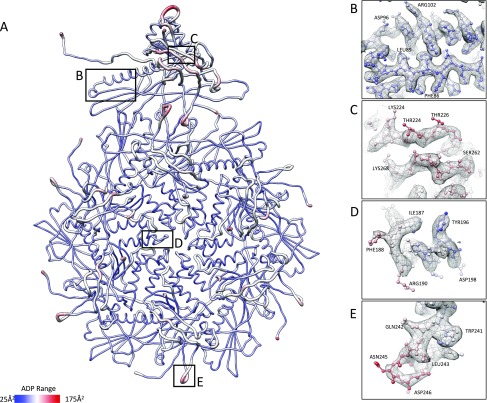
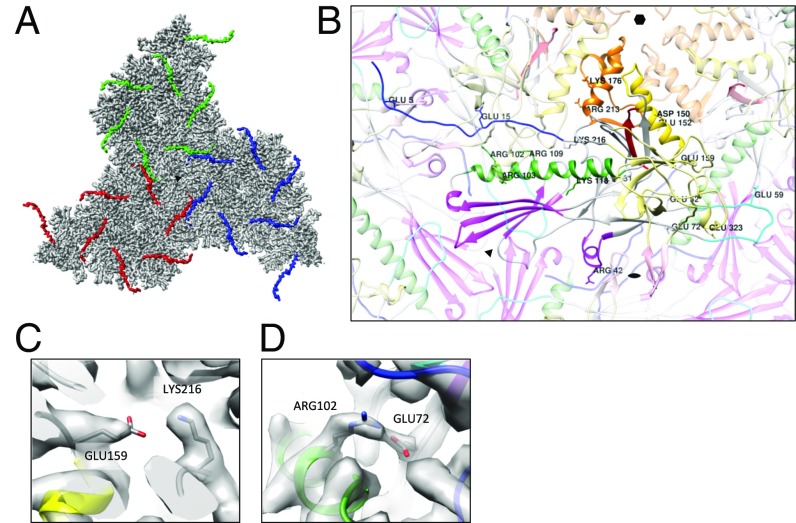
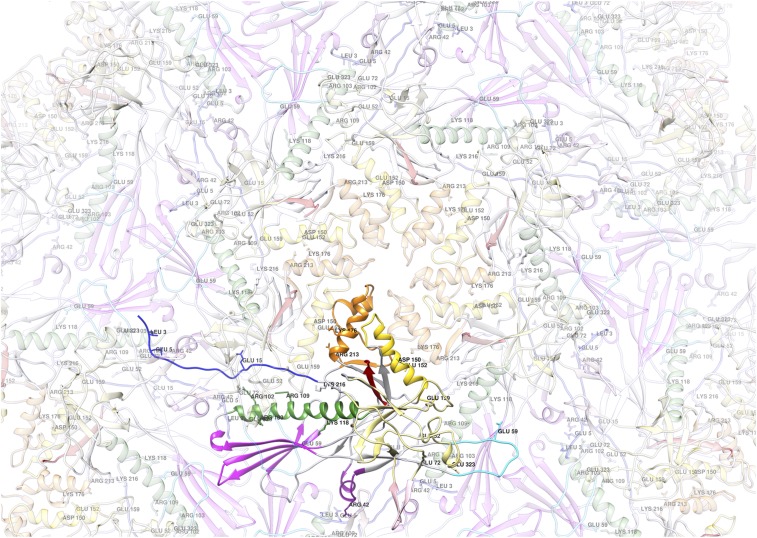
Electron cryomicroscopy (cryo-EM) has been used to determine the atomic coordinates (models) from density maps of biological assemblies. These models can be assessed by their overall fit to the experimental data and stereochemical information. However, these models do not annotate the actual density values of the atoms nor their positional uncertainty. Here, we introduce a computational procedure to derive an atomic model from a cryo-EM map with annotated metadata. The accuracy of such a model is validated by a faithful replication of the experimental cryo-EM map computed using the coordinates and associated metadata. The functional interpretation of any structural features in the model and its utilization for future studies can be made in the context of its measure of uncertainty. We applied this protocol to the 3.3-Å map of the mature P22 bacteriophage capsid, a large and complex macromolecular assembly. With this protocol, we identify and annotate previously undescribed molecular interactions between capsid subunits that are crucial to maintain stability in the absence of cementing proteins or cross-linking, as occur in other bacteriophages.
Keywords: P22; annotation; cryo-EM; model; structure.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




Similar articles
-
Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta.Elife. 2016 Sep 26;5:e17219. doi: 10.7554/eLife.17219. Elife. 2016. PMID: 27669148 Free PMC article.
-
Cryo-EM of macromolecular assemblies at near-atomic resolution.Nat Protoc. 2010 Sep;5(10):1697-708. doi: 10.1038/nprot.2010.126. Epub 2010 Sep 30. Nat Protoc. 2010. PMID: 20885381 Free PMC article.
-
Improving the Accuracy of Fitted Atomic Models in Cryo-EM Density Maps of Protein Assemblies Using Evolutionary Information from Aligned Homologous Proteins.Methods Mol Biol. 2016;1415:193-209. doi: 10.1007/978-1-4939-3572-7_10. Methods Mol Biol. 2016. PMID: 27115634
-
Single-particle cryo-electron microscopy of macromolecular complexes.Microscopy (Oxf). 2016 Feb;65(1):9-22. doi: 10.1093/jmicro/dfv366. Epub 2015 Nov 25. Microscopy (Oxf). 2016. PMID: 26611544 Free PMC article. Review.
-
Near-atomic-resolution cryo-EM for molecular virology.Curr Opin Virol. 2011 Aug;1(2):110-7. doi: 10.1016/j.coviro.2011.05.019. Curr Opin Virol. 2011. PMID: 21845206 Free PMC article. Review.
Cited by
-
Raman Multi-Omic Snapshot and Statistical Validation of Structural Differences between Herpes Simplex Type I and Epstein-Barr Viruses.Int J Mol Sci. 2023 Oct 25;24(21):15567. doi: 10.3390/ijms242115567. Int J Mol Sci. 2023. PMID: 37958551 Free PMC article.
-
Molecular exclusion limits for diffusion across a porous capsid.Nat Commun. 2021 May 18;12(1):2903. doi: 10.1038/s41467-021-23200-1. Nat Commun. 2021. PMID: 34006828 Free PMC article.
-
Principles for enhancing virus capsid capacity and stability from a thermophilic virus capsid structure.Nat Commun. 2019 Oct 2;10(1):4471. doi: 10.1038/s41467-019-12341-z. Nat Commun. 2019. PMID: 31578335 Free PMC article.
-
Structural basis for anthrax toxin receptor 1 recognition by Seneca Valley Virus.Proc Natl Acad Sci U S A. 2018 Nov 13;115(46):E10934-E10940. doi: 10.1073/pnas.1810664115. Epub 2018 Oct 31. Proc Natl Acad Sci U S A. 2018. PMID: 30381454 Free PMC article.
-
Simulation and Machine Learning Methods for Ion-Channel Structure Determination, Mechanistic Studies and Drug Design.Front Pharmacol. 2022 Jun 28;13:939555. doi: 10.3389/fphar.2022.939555. eCollection 2022. Front Pharmacol. 2022. PMID: 35837274 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
