

FANCD2 Binds Human Papillomavirus Genomes and Associates with a Distinct Set of DNA Repair Proteins to Regulate Viral Replication
- PMID: 28196964
- PMCID: PMC5312087
- DOI: 10.1128/mBio.02340-16
FANCD2 Binds Human Papillomavirus Genomes and Associates with a Distinct Set of DNA Repair Proteins to Regulate Viral Replication
Abstract
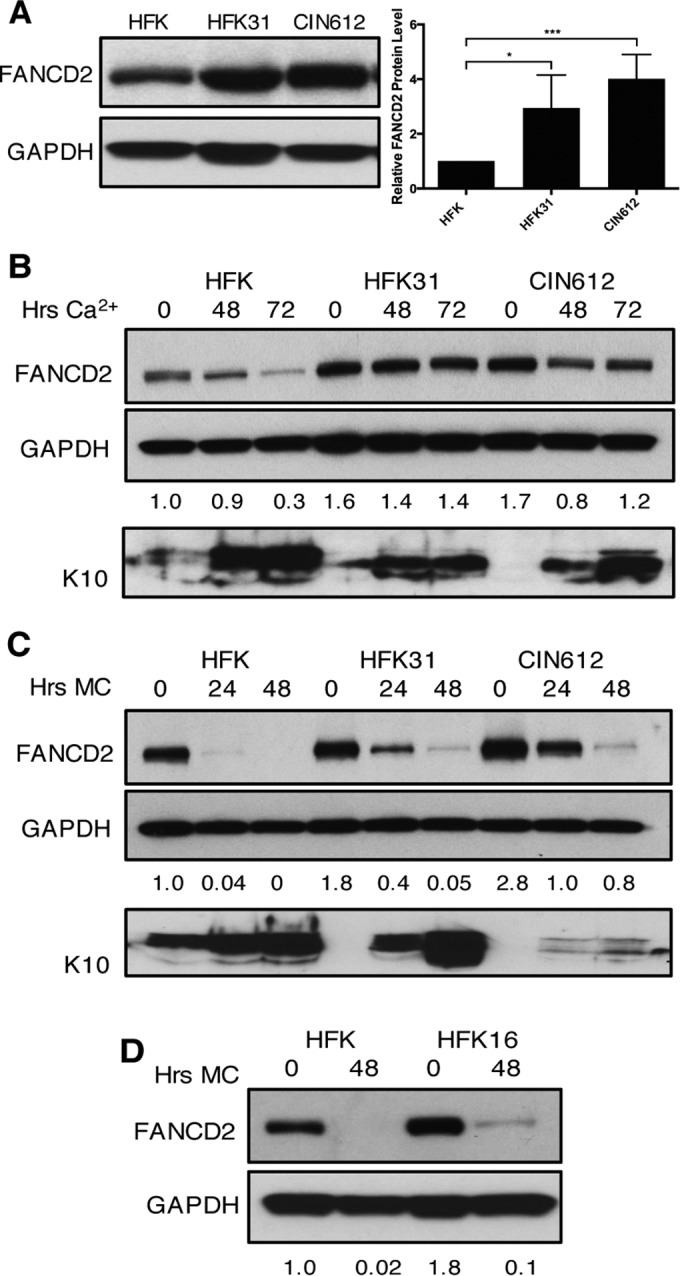
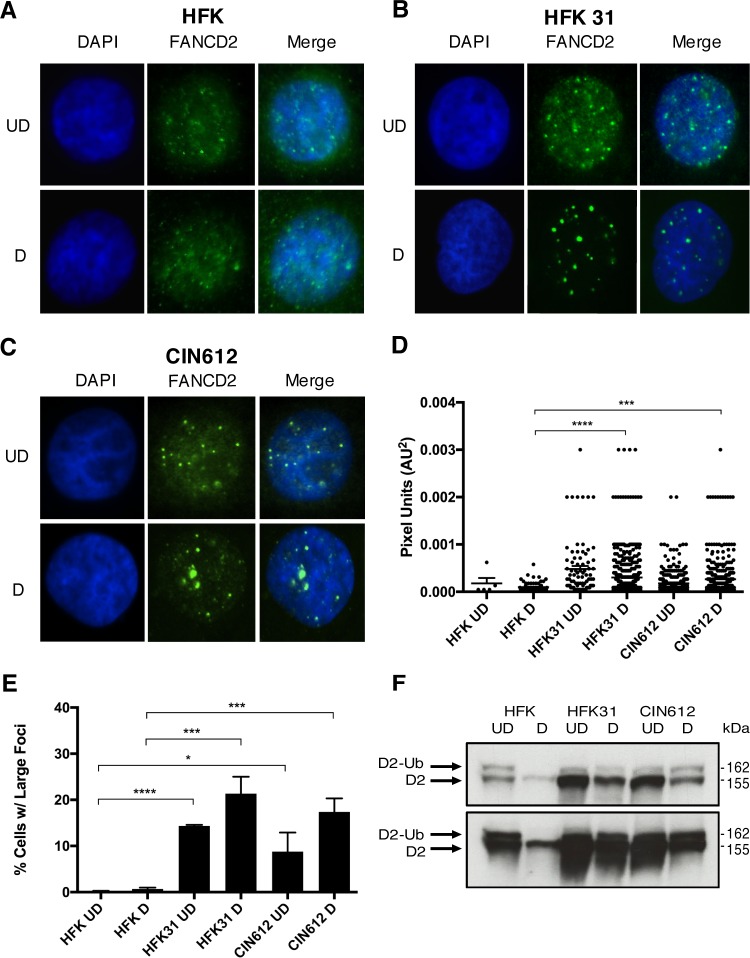
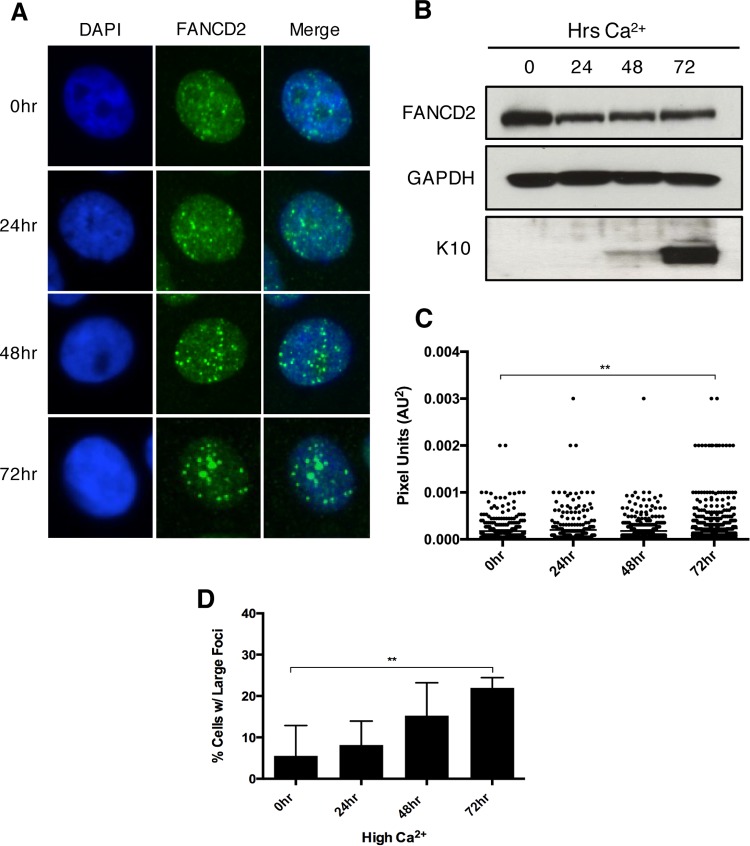
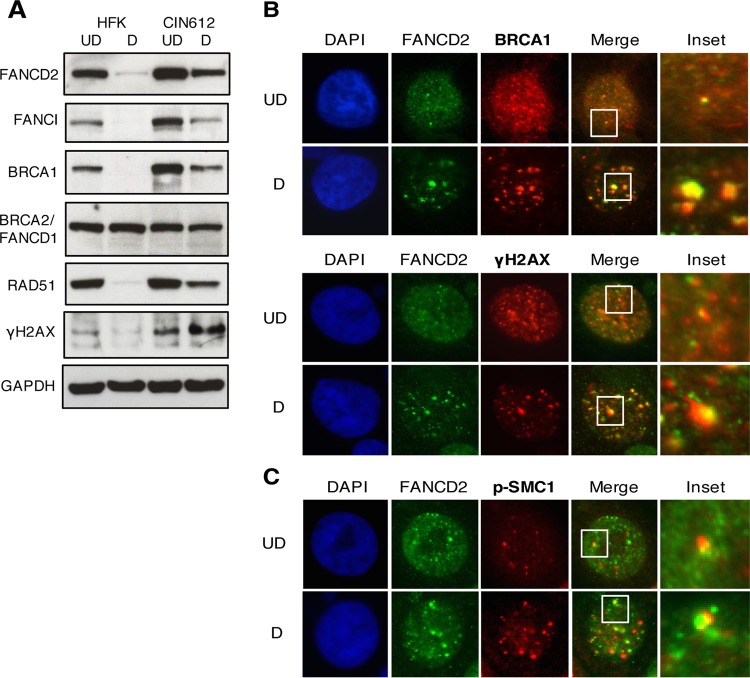
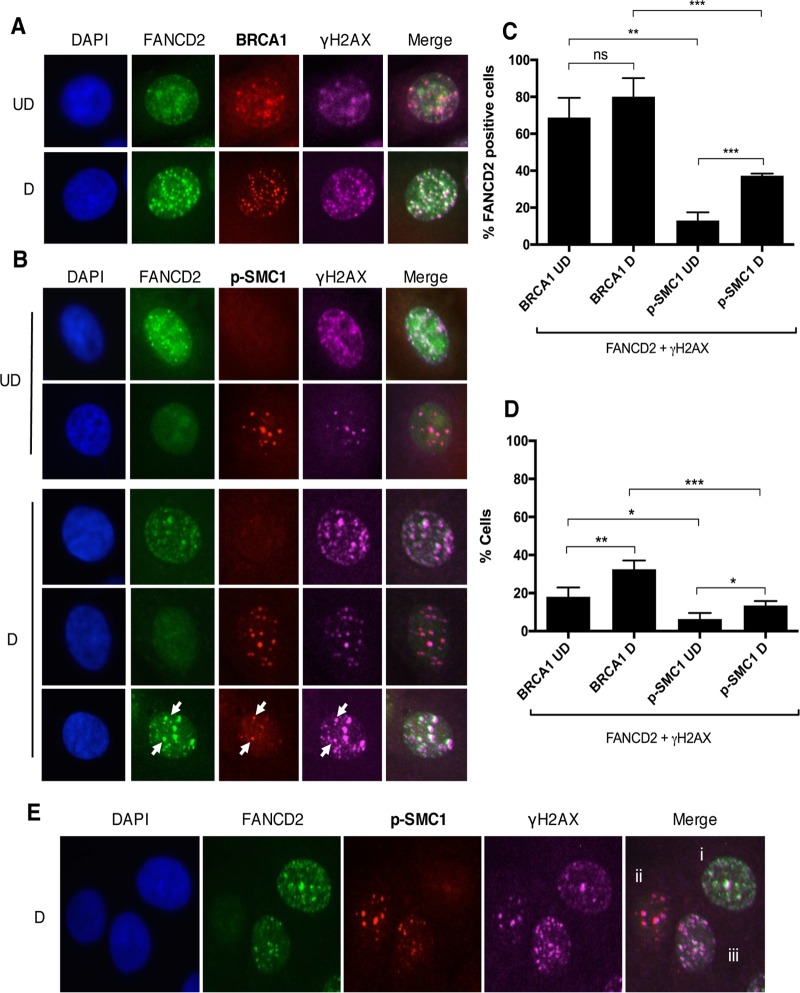
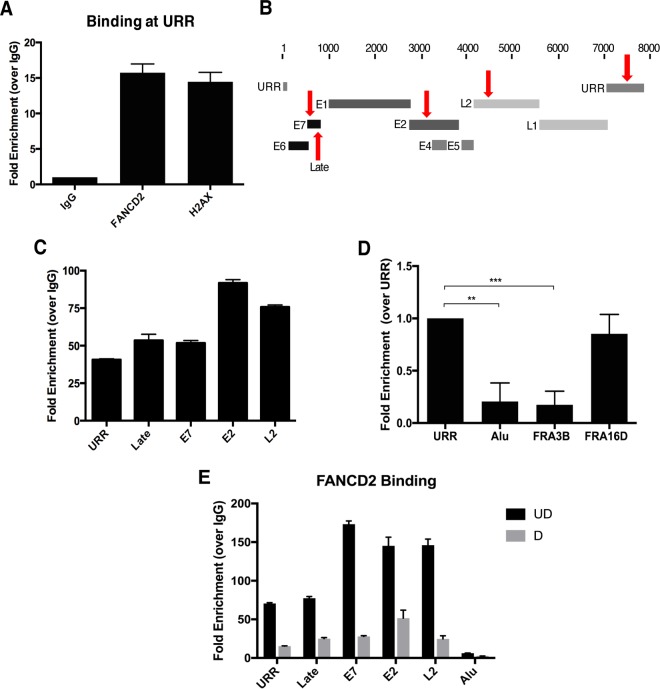
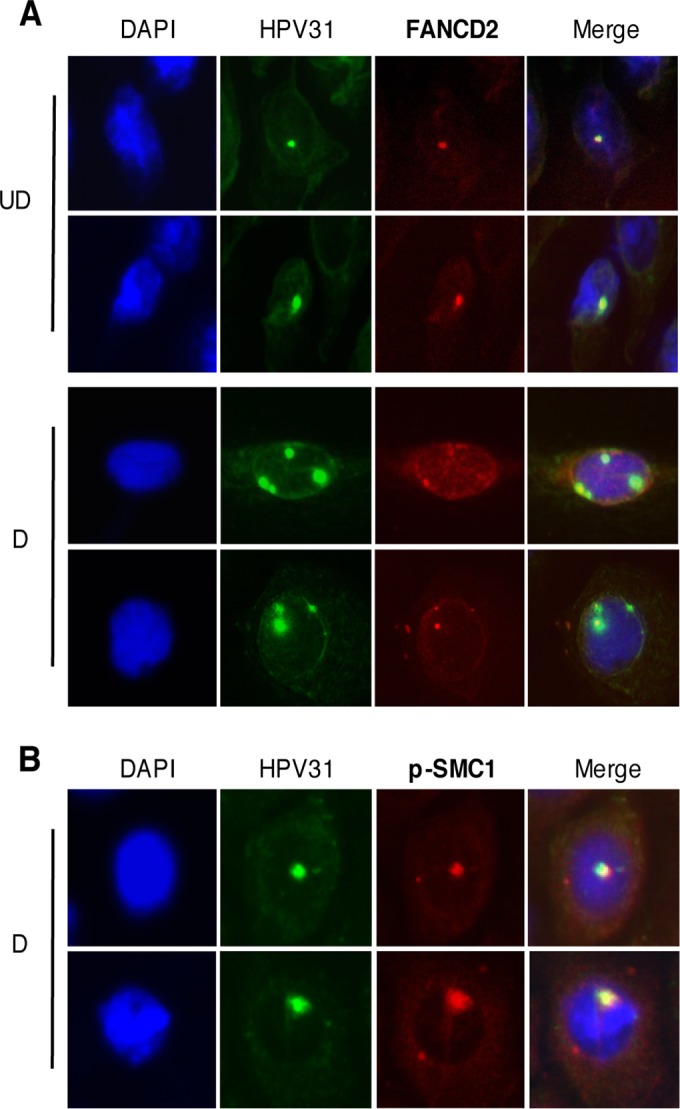
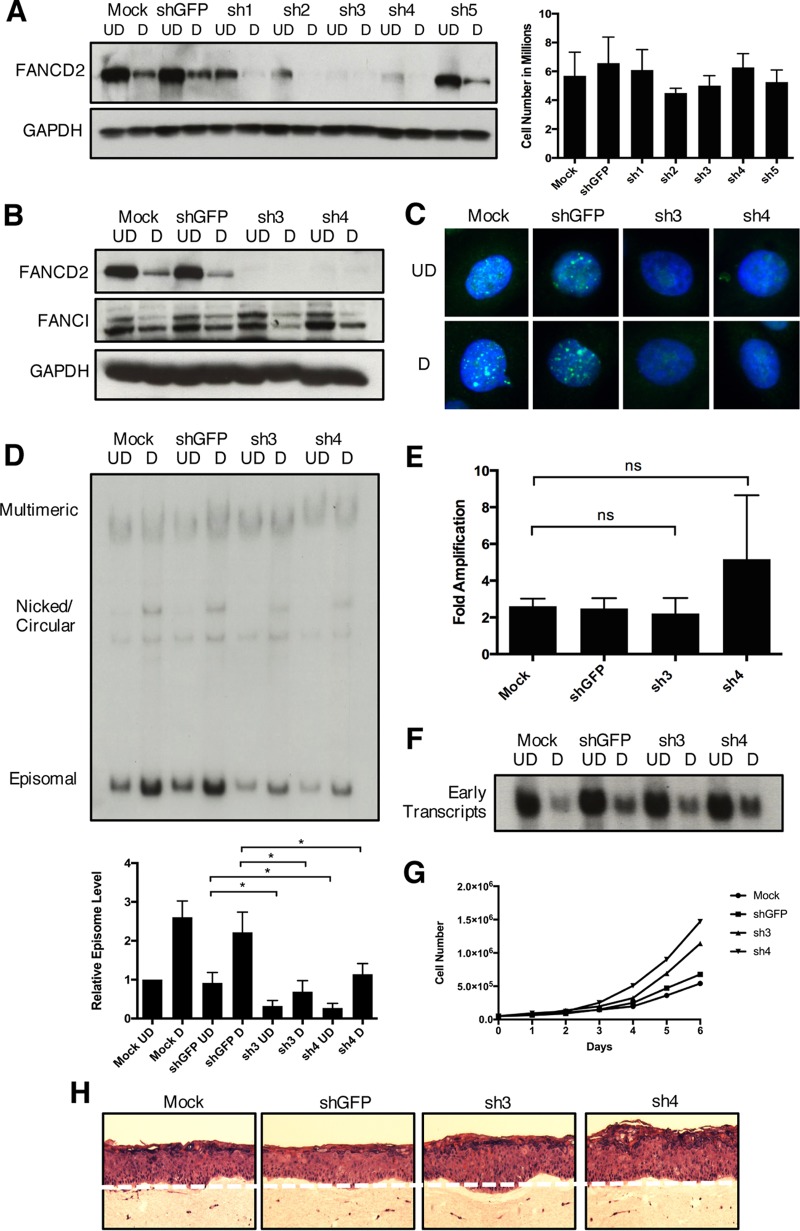
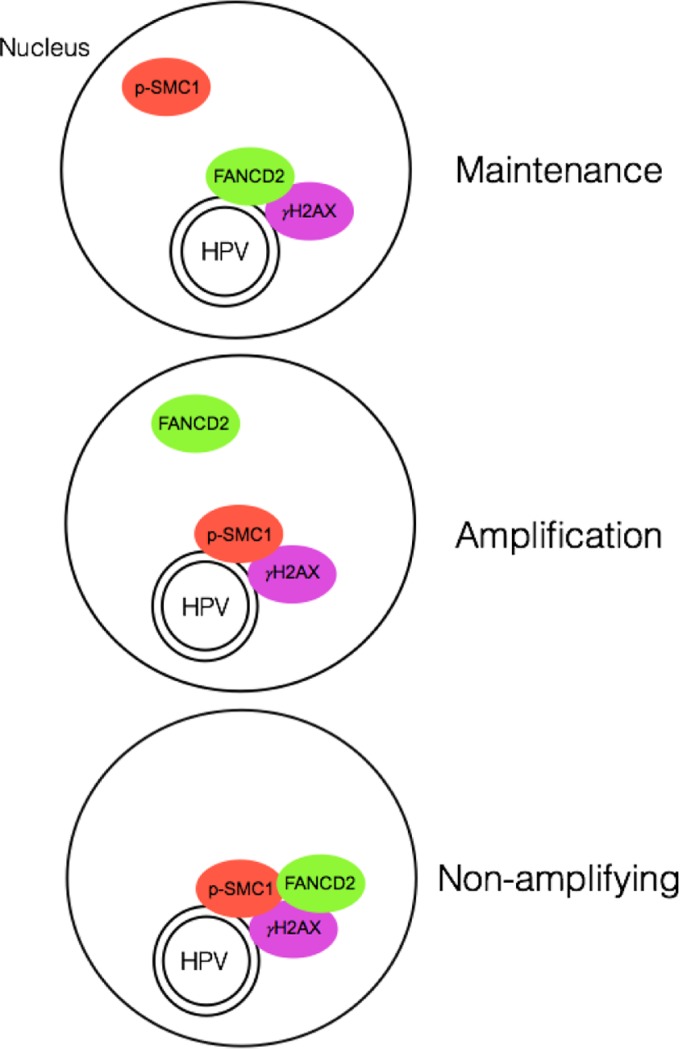
The life cycle of human papillomavirus (HPV) is dependent on the differentiation state of its host cell. HPV genomes are maintained as low-copy episomes in basal epithelial cells and amplified to thousands of copies per cell in differentiated layers. Replication of high-risk HPVs requires the activation of the ataxia telangiectasia-mutated (ATM) and ATM and Rad3-related (ATR) DNA repair pathways. The Fanconi anemia (FA) pathway is a part of the DNA damage response and mediates cross talk between the ATM and ATR pathways. Our studies show that HPV activates the FA pathway, leading to the accumulation of a key regulatory protein, FANCD2, in large nuclear foci. These HPV-dependent foci colocalize with a distinct population of DNA repair proteins, including ATM components γH2AX and BRCA1, but infrequently with p-SMC1, which is required for viral genome amplification in differentiated cells. Furthermore, FANCD2 is found at viral replication foci, where it is preferentially recruited to viral genomes compared to cellular chromosomes and is required for maintenance of HPV episomes in undifferentiated cells. These findings identify FANCD2 as an important regulator of HPV replication and provide insight into the role of the DNA damage response in the differentiation-dependent life cycle of HPV.IMPORTANCE High-risk human papillomaviruses (HPVs) are the etiological agents of cervical cancer and are linked to the development of many other anogenital and oropharyngeal cancers. Identification of host cellular pathways involved in regulating the viral life cycle may be helpful in identifying treatments for HPV lesions. Mutations in genes of the Fanconi anemia (FA) DNA repair pathway lead to genomic instability in patients and a predisposition to HPV-associated malignancies. Our studies demonstrate that FA pathway component FANCD2 is recruited to HPV DNA, associates with members of the ATM DNA repair pathway, and is essential for the maintenance of viral episomes in basal epithelial cells. Disruption of the FA pathway may result in increased integration events and a higher incidence of HPV-related cancer. Our study identifies new links between HPV and the FA pathway that may help to identify new therapeutic targets for the treatment of existing HPV infections and cancers.
Copyright © 2017 Spriggs and Laimins.
Figures
Similar articles
-
STAT-5 Regulates Transcription of the Topoisomerase IIβ-Binding Protein 1 (TopBP1) Gene To Activate the ATR Pathway and Promote Human Papillomavirus Replication.mBio. 2015 Dec 22;6(6):e02006-15. doi: 10.1128/mBio.02006-15. mBio. 2015. PMID: 26695634 Free PMC article.
-
Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification.mBio. 2018 Feb 13;9(1):e00064-18. doi: 10.1128/mBio.00064-18. mBio. 2018. PMID: 29440569 Free PMC article.
-
The fanconi anemia pathway limits human papillomavirus replication.J Virol. 2012 Aug;86(15):8131-8. doi: 10.1128/JVI.00408-12. Epub 2012 May 23. J Virol. 2012. PMID: 22623785 Free PMC article.
-
Modulation of the DNA damage response during the life cycle of human papillomaviruses.Virus Res. 2017 Mar 2;231:41-49. doi: 10.1016/j.virusres.2016.11.006. Epub 2016 Nov 9. Virus Res. 2017. PMID: 27836727 Free PMC article. Review.
-
Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication.Viruses. 2017 Aug 18;9(8):232. doi: 10.3390/v9080232. Viruses. 2017. PMID: 28820495 Free PMC article. Review.
Cited by
-
Interaction of the Human Papillomavirus E1 Helicase with UAF1-USP1 Promotes Unidirectional Theta Replication of Viral Genomes.mBio. 2019 Mar 19;10(2):e00152-19. doi: 10.1128/mBio.00152-19. mBio. 2019. PMID: 30890612 Free PMC article.
-
Interactions among human papillomavirus proteins and host DNA repair factors differ during the viral life cycle and virus-induced tumorigenesis.mSphere. 2023 Dec 20;8(6):e0042723. doi: 10.1128/msphere.00427-23. Epub 2023 Oct 18. mSphere. 2023. PMID: 37850786 Free PMC article. Review.
-
Virus DNA Replication and the Host DNA Damage Response.Annu Rev Virol. 2018 Sep 29;5(1):141-164. doi: 10.1146/annurev-virology-092917-043534. Epub 2018 Jul 11. Annu Rev Virol. 2018. PMID: 29996066 Free PMC article. Review.
-
Parvovirus minute virus of mice interacts with sites of cellular DNA damage to establish and amplify its lytic infection.Elife. 2018 Jul 20;7:e37750. doi: 10.7554/eLife.37750. Elife. 2018. PMID: 30028293 Free PMC article.
-
Mechanisms of persistence by small DNA tumor viruses.Curr Opin Virol. 2018 Oct;32:71-79. doi: 10.1016/j.coviro.2018.09.002. Epub 2018 Oct 1. Curr Opin Virol. 2018. PMID: 30278284 Free PMC article. Review.
References
-
- Lee C, Laimins LA. 2007. The differentiation-dependent life cycle of human papillomaviruses in keratinocytes, p 45–68. In Dimaio D, Garcea D (ed), The papillomaviruses. Springer, New York, NY.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
