

Sex specific activation of the ERα axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS
- PMID: 28186560
- PMCID: PMC6075578
- DOI: 10.1093/hmg/ddx049
Sex specific activation of the ERα axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS
Abstract
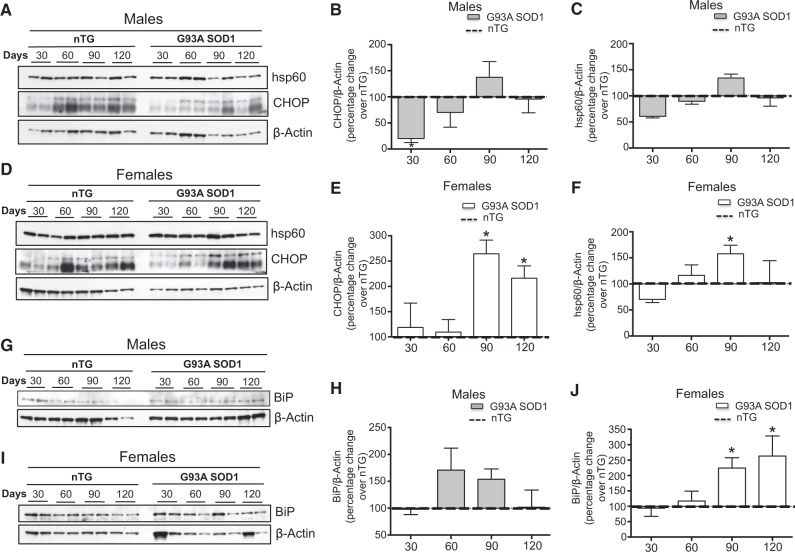
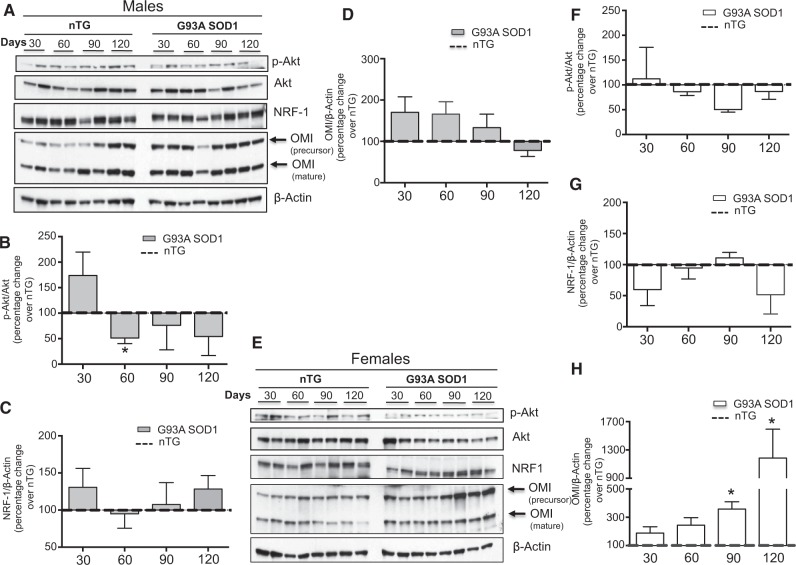
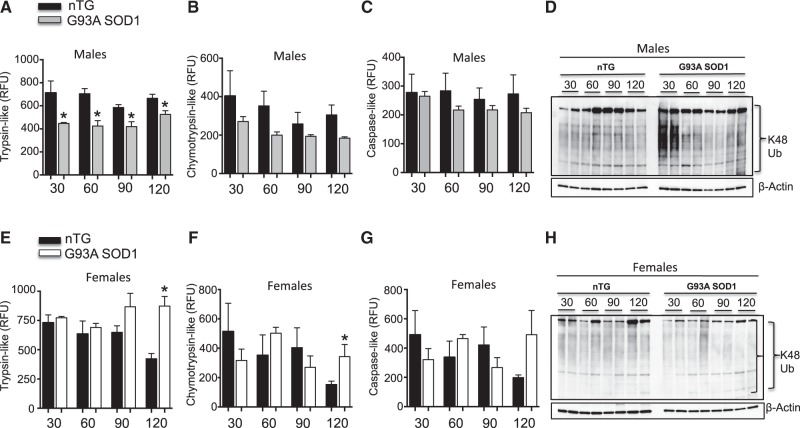
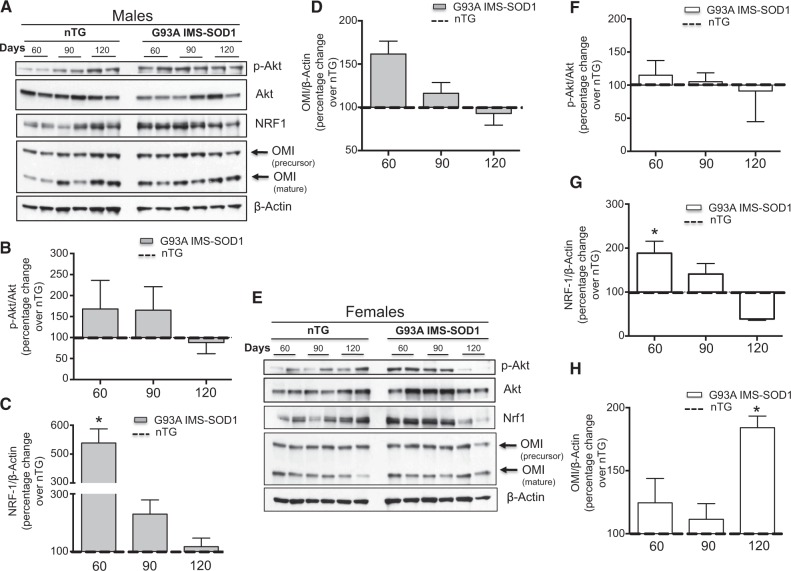
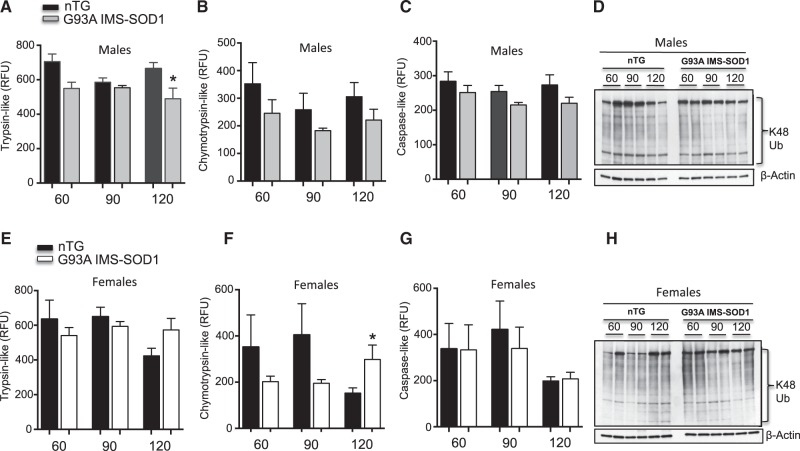
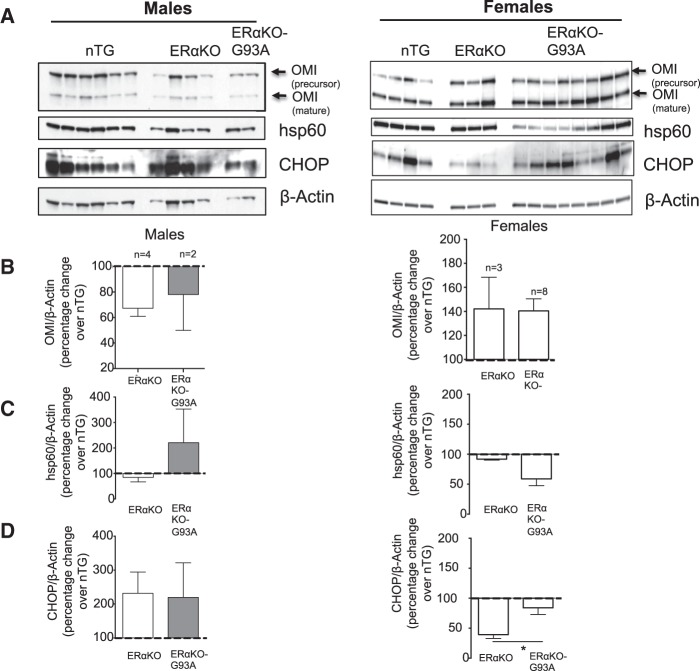
The mitochondrial unfolded protein response (UPRmt) is a transcriptional program aimed at restoring proteostasis in mitochondria. Upregulation of mitochondrial matrix proteases and heat shock proteins was initially described. Soon thereafter, a distinct UPRmt induced by misfolded proteins in the mitochondrial intermembrane space (IMS) and mediated by the estrogen receptor alpha (ERα), was found to upregulate the proteasome and the IMS protease OMI. However, the IMS-UPRmt was never studied in a neurodegenerative disease in vivo. Thus, we investigated the IMS-UPRmt in the G93A-SOD1 mouse model of familial ALS, since mutant SOD1 is known to accumulate in the IMS of neural tissue and cause mitochondrial dysfunction. As the ERα is most active in females, we postulated that a differential involvement of the IMS-UPRmt could be linked to the longer lifespan of females in the G93A-SOD1 mouse. We found a significant sex difference in the IMS-UPRmt, because the spinal cords of female, but not male, G93A-SOD1 mice showed elevation of OMI and proteasome activity. Then, using a mouse in which G93A-SOD1 was selectively targeted to the IMS, we demonstrated that the IMS-UPRmt could be specifically initiated by mutant SOD1 localized in the IMS. Furthermore, we showed that, in the absence of ERα, G93A-SOD1 failed to activate OMI and the proteasome, confirming the ERα dependence of the response. Taken together, these results demonstrate the IMS-UPRmt activation in SOD1 familial ALS, and suggest that sex differences in the disease phenotype could be linked to differential activation of the ERα axis of the IMS-UPRmt.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space.J Neurosci. 2011 Nov 2;31(44):15826-37. doi: 10.1523/JNEUROSCI.1965-11.2011. J Neurosci. 2011. PMID: 22049426 Free PMC article.
-
Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention.Neurobiol Dis. 2011 Jun;42(3):265-75. doi: 10.1016/j.nbd.2011.01.016. Epub 2011 Feb 3. Neurobiol Dis. 2011. PMID: 21296666
-
The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS).Hum Mol Genet. 2010 Sep 1;19(17):3440-56. doi: 10.1093/hmg/ddq257. Epub 2010 Jun 22. Hum Mol Genet. 2010. PMID: 20570967
-
Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space.Antioxid Redox Signal. 2010 Nov 1;13(9):1375-84. doi: 10.1089/ars.2010.3212. Antioxid Redox Signal. 2010. PMID: 20367259 Free PMC article. Review.
-
Transgenic mice with human mutant genes causing Parkinson's disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration.Rev Neurosci. 2007;18(2):115-36. doi: 10.1515/revneuro.2007.18.2.115. Rev Neurosci. 2007. PMID: 17593875 Review.
Cited by
-
Interplay between Mitochondrial Protein Import and Respiratory Complexes Assembly in Neuronal Health and Degeneration.Life (Basel). 2021 May 11;11(5):432. doi: 10.3390/life11050432. Life (Basel). 2021. PMID: 34064758 Free PMC article. Review.
-
Rab Geranylgeranyltransferase Subunit Beta as a Potential Indicator to Assess the Progression of Amyotrophic Lateral Sclerosis.Brain Sci. 2023 Oct 30;13(11):1531. doi: 10.3390/brainsci13111531. Brain Sci. 2023. PMID: 38002490 Free PMC article.
-
Mitochondrial stress response and myogenic differentiation.Front Cell Dev Biol. 2024 Apr 12;12:1381417. doi: 10.3389/fcell.2024.1381417. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 38681520 Free PMC article. Review.
-
A bird's eye view of mitochondrial unfolded protein response in cancer: mechanisms, progression and further applications.Cell Death Dis. 2024 Sep 11;15(9):667. doi: 10.1038/s41419-024-07049-y. Cell Death Dis. 2024. PMID: 39261452 Free PMC article. Review.
-
Mitohormesis Primes Tumor Invasion and Metastasis.Cell Rep. 2019 May 21;27(8):2292-2303.e6. doi: 10.1016/j.celrep.2019.04.095. Cell Rep. 2019. PMID: 31116976 Free PMC article.
References
-
- Rosen D.R. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 364, 362.. - PubMed
-
- Hervias I., Beal M.F., Manfredi G. (2006) Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve., 33, 598–608. - PubMed
-
- Gurney M.E., Pu H., Chiu A.Y., Dal Canto M.C., Polchow C.Y., Alexander D.D., Caliendo J., Hentati A., Kwon Y.W., Deng H.X., and., et al. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science, 264, 1772–1775. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
