

3D-e-Chem-VM: Structural Cheminformatics Research Infrastructure in a Freely Available Virtual Machine
- PMID: 28125221
- PMCID: PMC5342320
- DOI: 10.1021/acs.jcim.6b00686
3D-e-Chem-VM: Structural Cheminformatics Research Infrastructure in a Freely Available Virtual Machine
Abstract
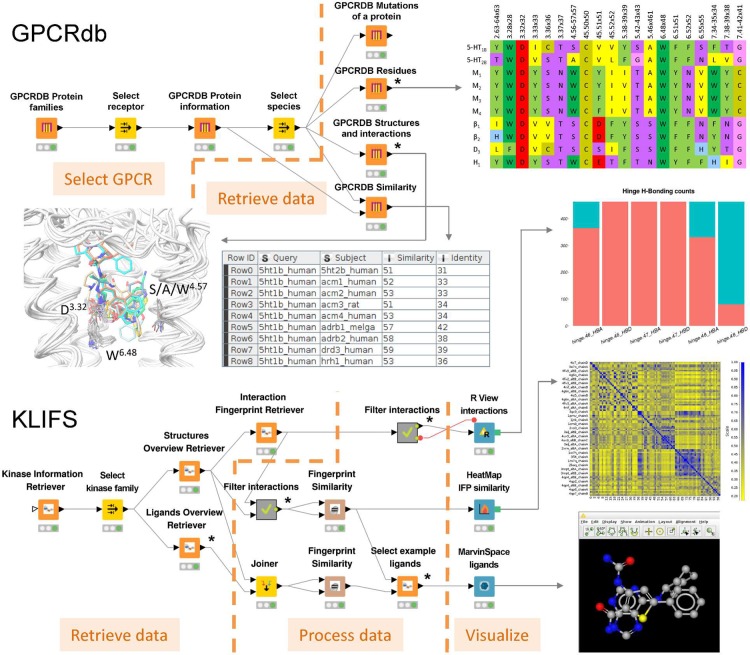
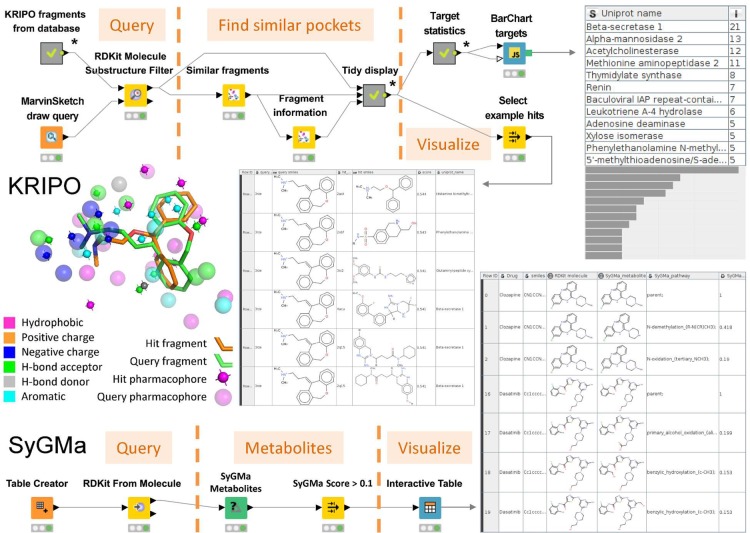
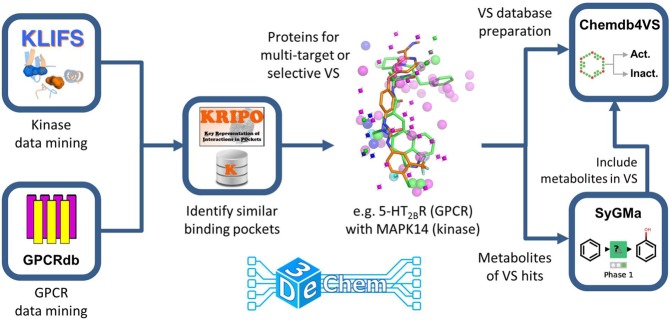
3D-e-Chem-VM is an open source, freely available Virtual Machine ( http://3d-e-chem.github.io/3D-e-Chem-VM/ ) that integrates cheminformatics and bioinformatics tools for the analysis of protein-ligand interaction data. 3D-e-Chem-VM consists of software libraries, and database and workflow tools that can analyze and combine small molecule and protein structural information in a graphical programming environment. New chemical and biological data analytics tools and workflows have been developed for the efficient exploitation of structural and pharmacological protein-ligand interaction data from proteomewide databases (e.g., ChEMBLdb and PDB), as well as customized information systems focused on, e.g., G protein-coupled receptors (GPCRdb) and protein kinases (KLIFS). The integrated structural cheminformatics research infrastructure compiled in the 3D-e-Chem-VM enables the design of new approaches in virtual ligand screening (Chemdb4VS), ligand-based metabolism prediction (SyGMa), and structure-based protein binding site comparison and bioisosteric replacement for ligand design (KRIPOdb).
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
3D-e-Chem: Structural Cheminformatics Workflows for Computer-Aided Drug Discovery.ChemMedChem. 2018 Mar 20;13(6):614-626. doi: 10.1002/cmdc.201700754. Epub 2018 Feb 14. ChemMedChem. 2018. PMID: 29337438 Free PMC article.
-
myChEMBL: a virtual machine implementation of open data and cheminformatics tools.Bioinformatics. 2014 Jan 15;30(2):298-300. doi: 10.1093/bioinformatics/btt666. Epub 2013 Nov 20. Bioinformatics. 2014. PMID: 24262214 Free PMC article.
-
Scipion-Chem: An Open Platform for Virtual Drug Screening.J Chem Inf Model. 2023 Dec 25;63(24):7873-7885. doi: 10.1021/acs.jcim.3c01085. Epub 2023 Dec 5. J Chem Inf Model. 2023. PMID: 38052452 Free PMC article.
-
From cheminformatics to structure-based design: Web services and desktop applications based on the NAOMI library.J Biotechnol. 2017 Nov 10;261:207-214. doi: 10.1016/j.jbiotec.2017.06.004. Epub 2017 Jun 11. J Biotechnol. 2017. PMID: 28610996 Review.
-
Molecular interaction fingerprint approaches for GPCR drug discovery.Curr Opin Pharmacol. 2016 Oct;30:59-68. doi: 10.1016/j.coph.2016.07.007. Epub 2016 Jul 29. Curr Opin Pharmacol. 2016. PMID: 27479316 Review.
Cited by
-
Exploring Selectivity of Multikinase Inhibitors across the Human Kinome.ACS Omega. 2018 Jan 31;3(1):1147-1153. doi: 10.1021/acsomega.7b01960. Epub 2018 Jan 26. ACS Omega. 2018. PMID: 30221217 Free PMC article.
-
Towards reproducible computational drug discovery.J Cheminform. 2020 Jan 28;12(1):9. doi: 10.1186/s13321-020-0408-x. J Cheminform. 2020. PMID: 33430992 Free PMC article. Review.
-
KLIFS: an overhaul after the first 5 years of supporting kinase research.Nucleic Acids Res. 2021 Jan 8;49(D1):D562-D569. doi: 10.1093/nar/gkaa895. Nucleic Acids Res. 2021. PMID: 33084889 Free PMC article.
-
3D-e-Chem: Structural Cheminformatics Workflows for Computer-Aided Drug Discovery.ChemMedChem. 2018 Mar 20;13(6):614-626. doi: 10.1002/cmdc.201700754. Epub 2018 Feb 14. ChemMedChem. 2018. PMID: 29337438 Free PMC article.
-
Kinase inhibitors: the road ahead.Nat Rev Drug Discov. 2018 May;17(5):353-377. doi: 10.1038/nrd.2018.21. Epub 2018 Mar 16. Nat Rev Drug Discov. 2018. PMID: 29545548 Review.
References
-
- RDKit. http://www.rdkit.org.
-
- Jmol. http://jmol.sourceforge.net/.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
