

Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds
- PMID: 28124989
- PMCID: PMC5297856
- DOI: 10.3390/ijms18010227
Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds
Abstract
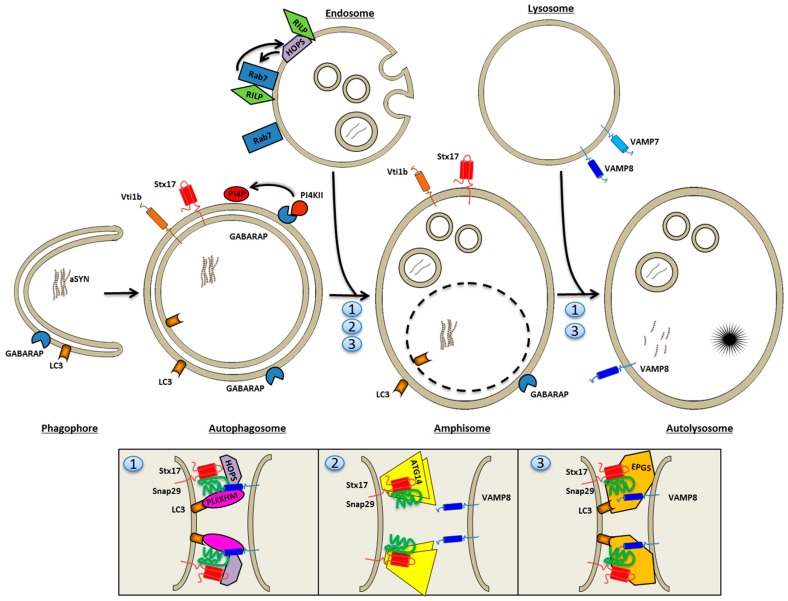
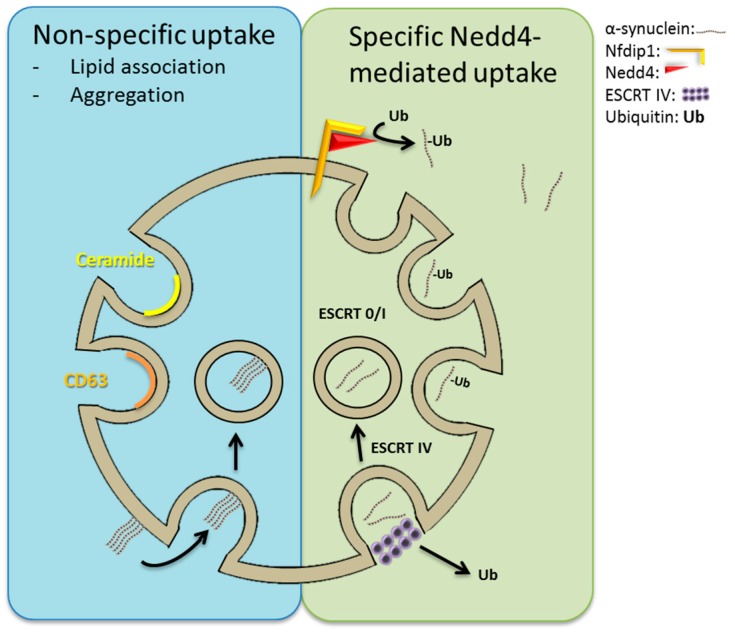
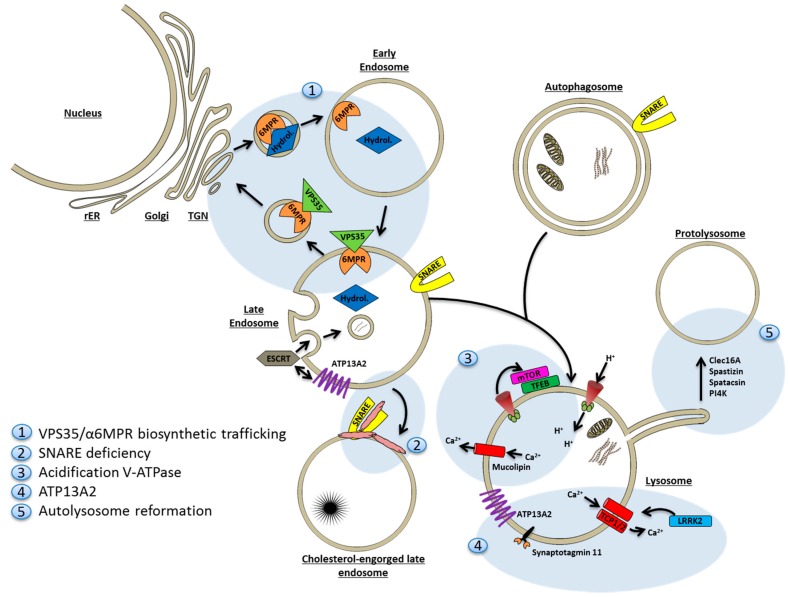
A mechanistic link between neuron-to-neuron transmission of secreted amyloid and propagation of protein malconformation cytopathology and disease has recently been uncovered in animal models. An enormous interest in the unconventional secretion of amyloids from neurons has followed. Amphisomes and late endosomes are the penultimate maturation products of the autophagosomal and endosomal pathways, respectively, and normally fuse with lysosomes for degradation. However, under conditions of perturbed membrane trafficking and/or lysosomal deficiency, prelysosomal compartments may instead fuse with the plasma membrane to release any contained amyloid. After a brief introduction to the endosomal and autophagosomal pathways, we discuss the evidence for autophagosomal secretion (exophagy) of amyloids, with a comparative emphasis on Aβ1-42 and α-synuclein, as luminal and cytosolic amyloids, respectively. The ESCRT-mediated import of cytosolic amyloid into late endosomal exosomes, a known vehicle of transmission of macromolecules between cells, is also reviewed. Finally, mechanisms of lysosomal dysfunction, deficiency, and exocytosis are exemplified in the context of genetically identified risk factors, mainly for Parkinson's disease. Exocytosis of prelysosomal or lysosomal organelles is a last resort for clearance of cytotoxic material and alleviates cytopathy. However, they also represent a vehicle for the concentration, posttranslational modification, and secretion of amyloid seeds.
Keywords: autophagosomes; late endosomes; neurodegeneration; unconventional secretion; α-synuclein.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion.J Biol Chem. 2013 Jun 14;288(24):17313-35. doi: 10.1074/jbc.M112.401174. Epub 2013 Apr 29. J Biol Chem. 2013. PMID: 23629650 Free PMC article.
-
Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular.Int J Mol Sci. 2020 Apr 8;21(7):2576. doi: 10.3390/ijms21072576. Int J Mol Sci. 2020. PMID: 32276321 Free PMC article. Review.
-
Understanding amphisomes.Biochem J. 2021 May 28;478(10):1959-1976. doi: 10.1042/BCJ20200917. Biochem J. 2021. PMID: 34047789 Free PMC article. Review.
-
The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein.PLoS One. 2011;6(12):e29460. doi: 10.1371/journal.pone.0029460. Epub 2011 Dec 22. PLoS One. 2011. PMID: 22216284 Free PMC article.
-
Emerging autophagic endo-lysosomal targets in the management of Parkinson's disease.Rev Neurol (Paris). 2024 Jun;180(6):477-485. doi: 10.1016/j.neurol.2023.07.007. Epub 2023 Aug 14. Rev Neurol (Paris). 2024. PMID: 37586941 Review.
Cited by
-
Pathogenic Mutations Differentially Regulate Cell-to-Cell Transmission of α-Synuclein.Front Cell Neurosci. 2020 Jun 12;14:159. doi: 10.3389/fncel.2020.00159. eCollection 2020. Front Cell Neurosci. 2020. PMID: 32595456 Free PMC article.
-
LGALS3 (galectin 3) mediates an unconventional secretion of SNCA/α-synuclein in response to lysosomal membrane damage by the autophagic-lysosomal pathway in human midbrain dopamine neurons.Autophagy. 2022 May;18(5):1020-1048. doi: 10.1080/15548627.2021.1967615. Epub 2021 Oct 6. Autophagy. 2022. PMID: 34612142 Free PMC article.
-
Regulation of protein homeostasis by unconventional protein secretion in mammalian cells.Semin Cell Dev Biol. 2018 Nov;83:29-35. doi: 10.1016/j.semcdb.2018.03.006. Epub 2018 Mar 22. Semin Cell Dev Biol. 2018. PMID: 29549062 Free PMC article. Review.
-
Maturation and Clearance of Autophagosomes in Neurons Depends on a Specific Cysteine Protease Isoform, ATG-4.2.Dev Cell. 2019 Apr 22;49(2):251-266.e8. doi: 10.1016/j.devcel.2019.02.013. Epub 2019 Mar 14. Dev Cell. 2019. PMID: 30880001 Free PMC article.
-
Exosomal biomarkers in Down syndrome and Alzheimer's disease.Free Radic Biol Med. 2018 Jan;114:110-121. doi: 10.1016/j.freeradbiomed.2017.08.028. Epub 2017 Sep 5. Free Radic Biol Med. 2018. PMID: 28882786 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
