

RUNX3 is oncogenic in natural killer/T-cell lymphoma and is transcriptionally regulated by MYC
- PMID: 28119527
- PMCID: PMC5629367
- DOI: 10.1038/leu.2017.40
RUNX3 is oncogenic in natural killer/T-cell lymphoma and is transcriptionally regulated by MYC
Abstract
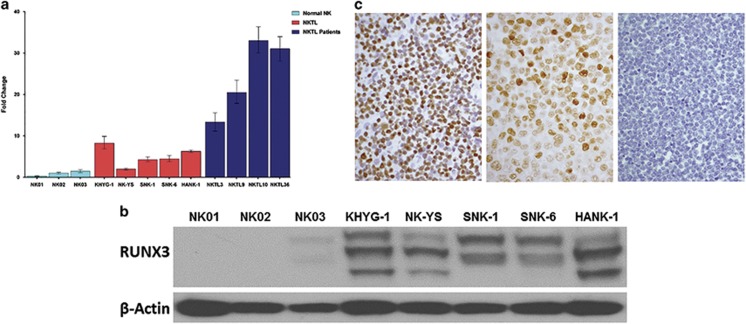
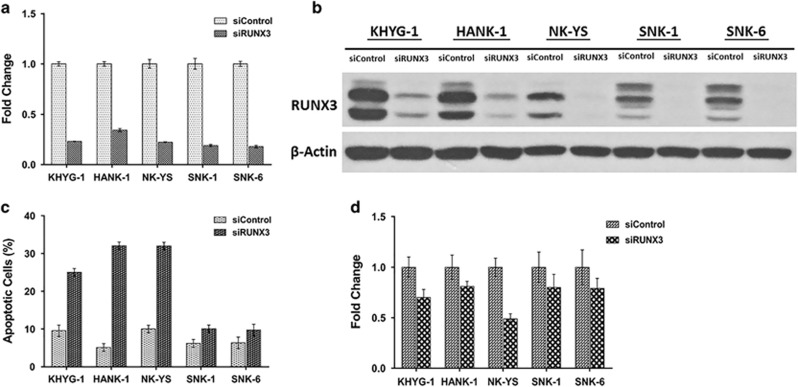
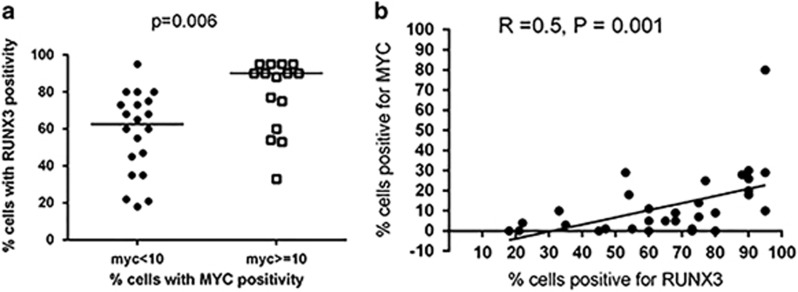
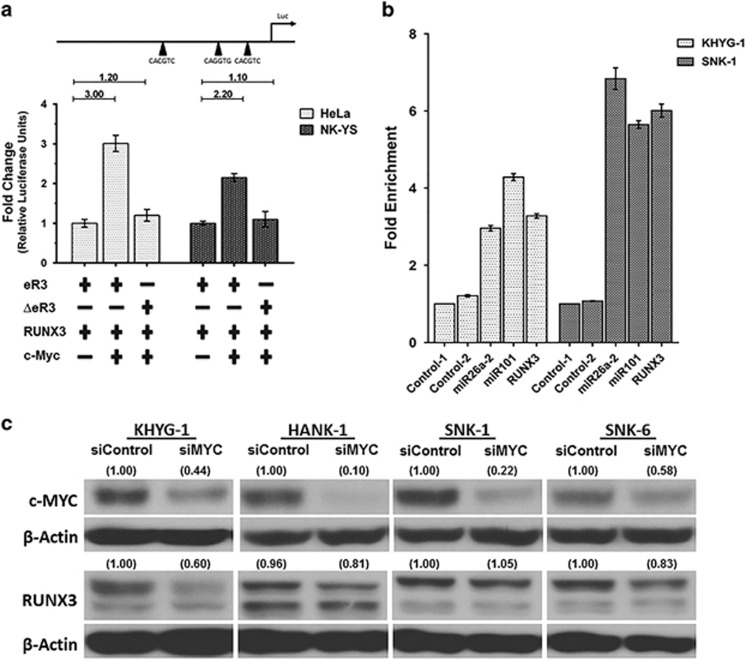
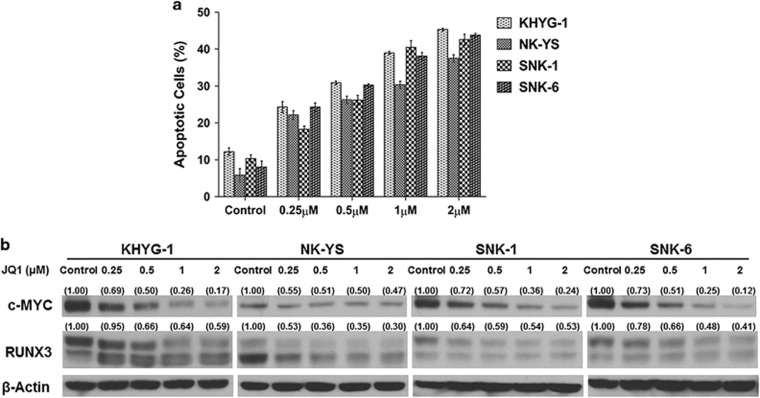
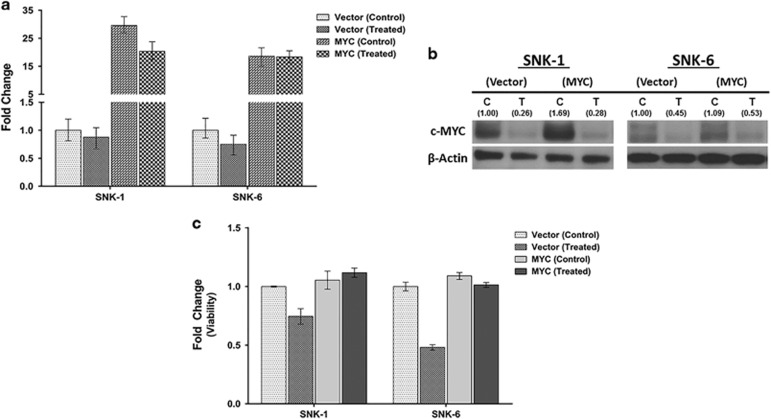
RUNX3, runt-domain transcription factor, is a master regulator of gene expression in major developmental pathways. It acts as a tumor suppressor in many cancers but is oncogenic in certain tumors. We observed upregulation of RUNX3 mRNA and protein expression in nasal-type extranodal natural killer (NK)/T-cell lymphoma (NKTL) patient samples and NKTL cell lines compared to normal NK cells. RUNX3 silenced NKTL cells showed increased apoptosis and reduced cell proliferation. Potential binding sites for MYC were identified in the RUNX3 enhancer region. Chromatin immunoprecipitation-quantitative PCR revealed binding activity between MYC and RUNX3. Co-transfection of the MYC expression vector with RUNX3 enhancer reporter plasmid resulted in activation of RUNX3 enhancer indicating that MYC positively regulates RUNX3 transcription in NKTL cell lines. Treatment with a small-molecule MYC inhibitor (JQ1) caused significant downregulation of MYC and RUNX3, leading to apoptosis in NKTL cells. The growth inhibition resulting from depletion of MYC by JQ1 was rescued by ectopic MYC expression. In summary, our study identified RUNX3 overexpression in NKTL with functional oncogenic properties. We further delineate that MYC may be an important upstream driver of RUNX3 upregulation and since MYC is upregulated in NKTL, further study on the employment of MYC inhibition as a therapeutic strategy is warranted.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Activated oncogenic pathways and therapeutic targets in extranodal nasal-type NK/T cell lymphoma revealed by gene expression profiling.J Pathol. 2011 Mar;223(4):496-510. doi: 10.1002/path.2823. Epub 2011 Jan 5. J Pathol. 2011. PMID: 21294123
-
ATP-binding cassette sub-family C member 4 (ABCC4) is overexpressed in human NK/T-cell lymphoma and regulates chemotherapy sensitivity: Potential as a functional therapeutic target.Leuk Res. 2015 Dec;39(12):1448-54. doi: 10.1016/j.leukres.2015.10.001. Epub 2015 Oct 15. Leuk Res. 2015. PMID: 26499190
-
Super-enhancer-driven TOX2 mediates oncogenesis in Natural Killer/T Cell Lymphoma.Mol Cancer. 2023 Apr 10;22(1):69. doi: 10.1186/s12943-023-01767-1. Mol Cancer. 2023. PMID: 37032358 Free PMC article.
-
Oncogenic RUNX3: A Link between p53 Deficiency and MYC Dysregulation.Mol Cells. 2020 Feb 29;43(2):176-181. doi: 10.14348/molcells.2019.0285. Mol Cells. 2020. PMID: 31991537 Free PMC article. Review.
-
Two faces of RUNX3 in myeloid transformation.Exp Hematol. 2021 May;97:14-20. doi: 10.1016/j.exphem.2021.02.005. Epub 2021 Feb 16. Exp Hematol. 2021. PMID: 33600870 Review.
Cited by
-
Epigenetic Mechanisms Dictating Eradication of Cancer by Natural Killer Cells.Trends Cancer. 2018 Aug;4(8):553-566. doi: 10.1016/j.trecan.2018.06.004. Epub 2018 Jul 3. Trends Cancer. 2018. PMID: 30064663 Free PMC article. Review.
-
The Genomics and Molecular Biology of Natural Killer/T-Cell Lymphoma: Opportunities for Translation.Int J Mol Sci. 2018 Jun 30;19(7):1931. doi: 10.3390/ijms19071931. Int J Mol Sci. 2018. PMID: 29966370 Free PMC article. Review.
-
Myeloid/natural killer (NK) cell precursor acute leukemia as a distinct leukemia type.Sci Adv. 2023 Dec 15;9(50):eadj4407. doi: 10.1126/sciadv.adj4407. Epub 2023 Dec 13. Sci Adv. 2023. PMID: 38091391 Free PMC article.
-
Disruption of c-MYC Binding and Chromosomal Looping Involving Genetic Variants Associated With Ankylosing Spondylitis Upstream of the RUNX3 Promoter.Front Genet. 2022 Jan 7;12:741867. doi: 10.3389/fgene.2021.741867. eCollection 2021. Front Genet. 2022. PMID: 35069677 Free PMC article.
-
Expression and Prognosis Analyses of Runt-Related Transcription Factor Family in Human Leukemia.Mol Ther Oncolytics. 2018 Dec 18;12:103-111. doi: 10.1016/j.omto.2018.12.008. eCollection 2019 Mar 29. Mol Ther Oncolytics. 2018. PMID: 30719500 Free PMC article.
References
-
- Ito Y. Oncogenic potential of the RUNX gene family: 'Overview'. Oncogene 2004; 23: 4198–4208. - PubMed
-
- Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Adv Cancer Res 2008; 99: 33–76. - PubMed
-
- Ito K, Liu Q, Salto-Tellez M, Yano T, Tada K, Ida H et al. RUNX3, a novel tumor suppressor, is frequently inactivated in gastric cancer by protein mislocalization. Cancer Res 2005; 65: 7743–7750. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
