

Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity
- PMID: 28103318
- PMCID: PMC5283768
- DOI: 10.1371/journal.ppat.1006136
Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity
Abstract
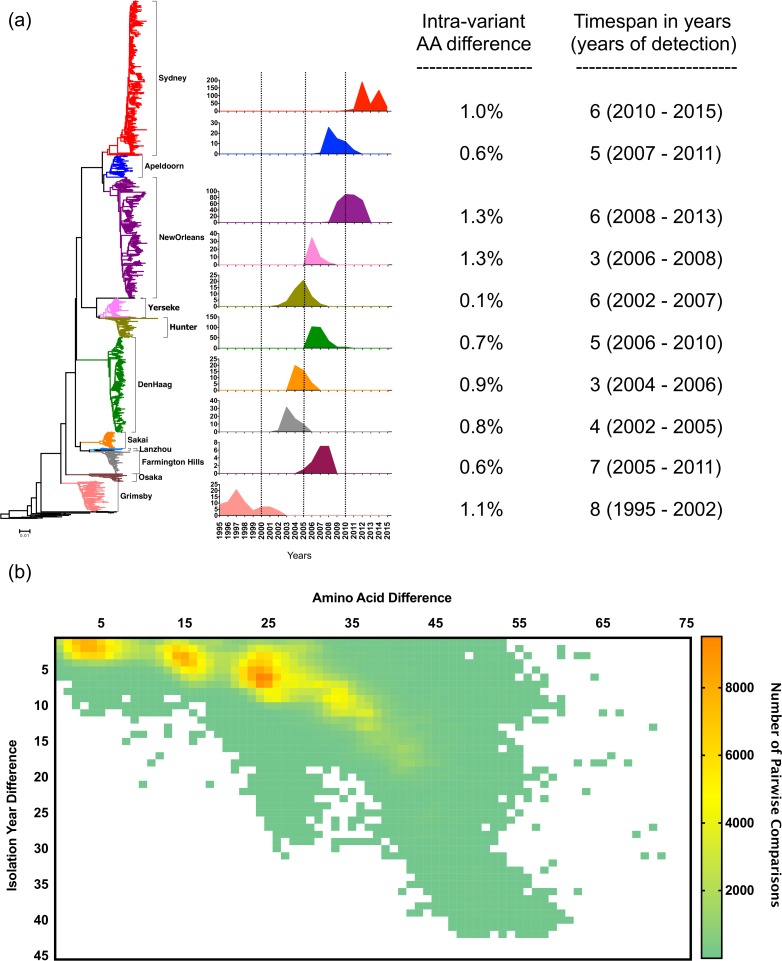
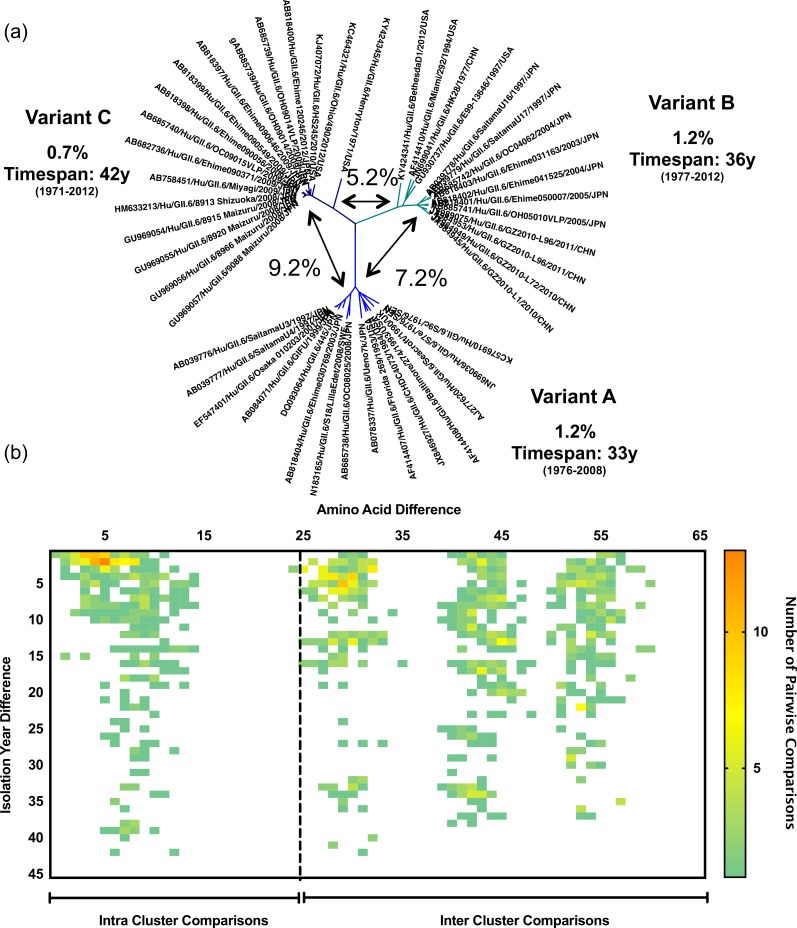
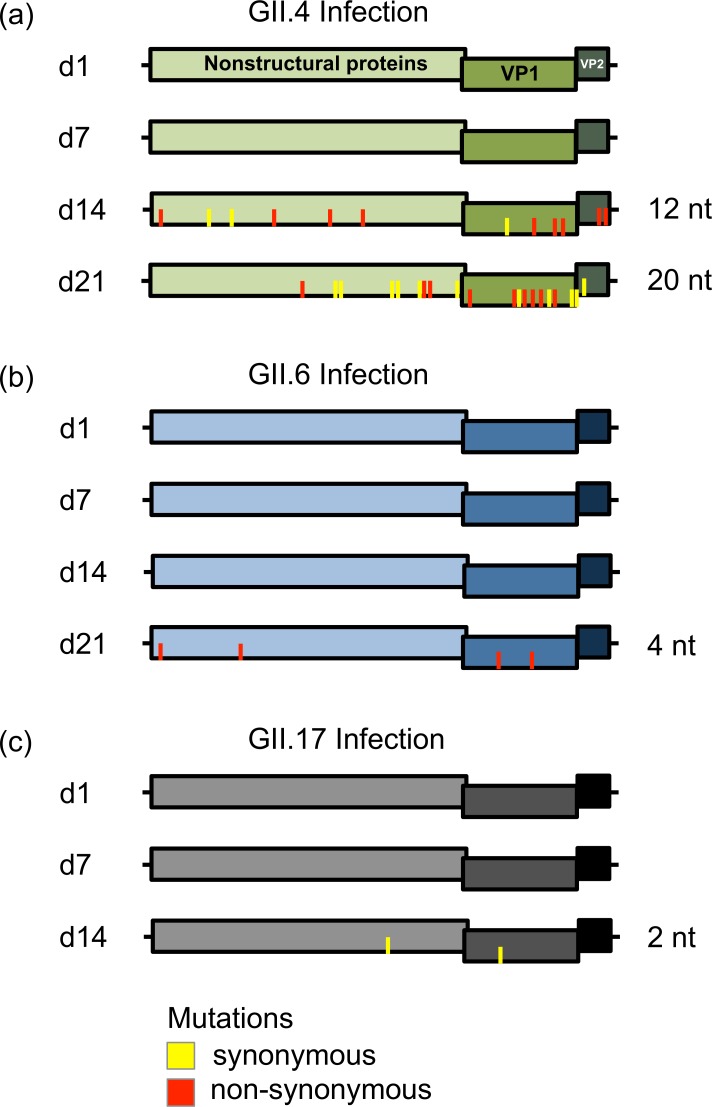
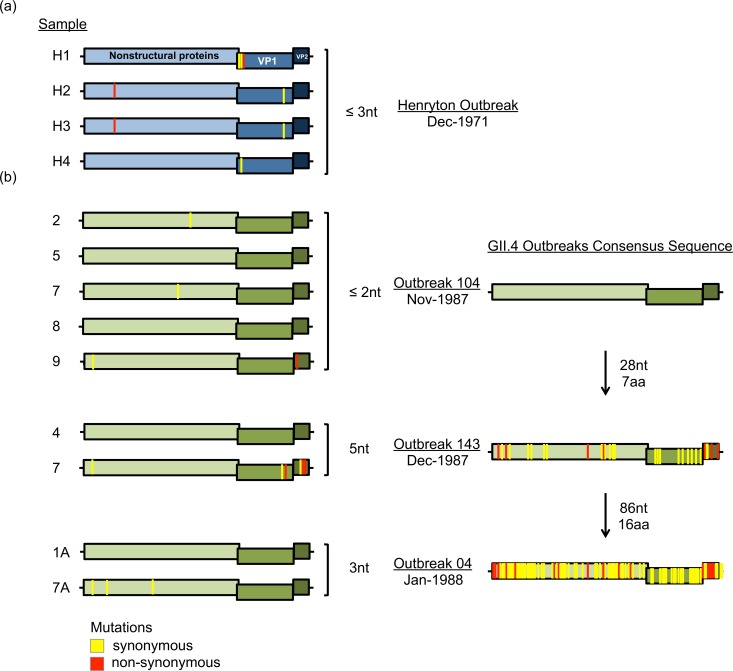
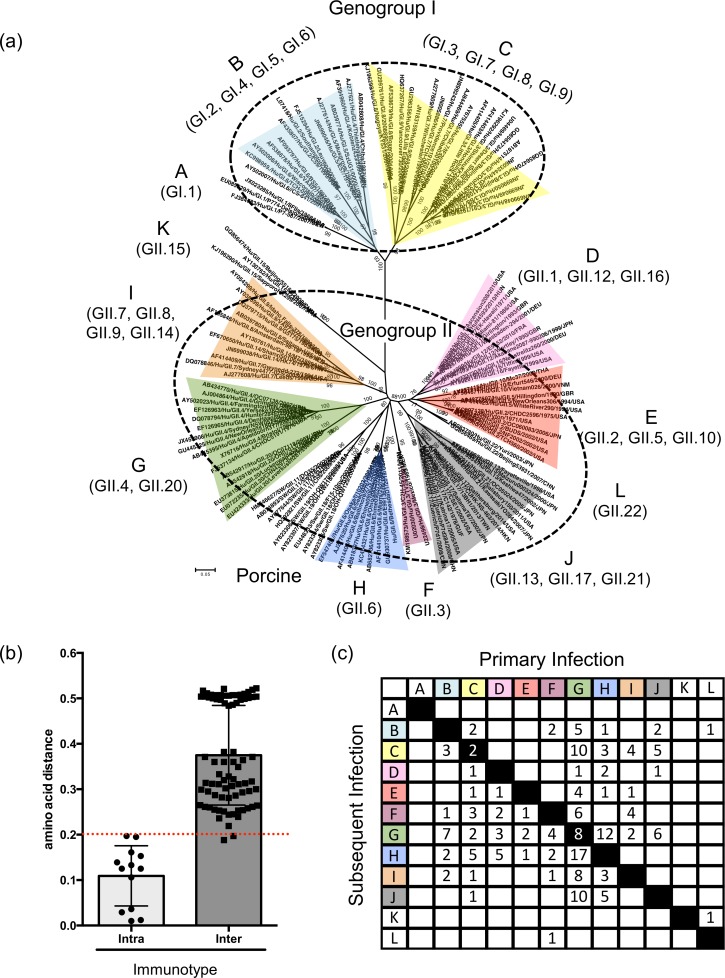
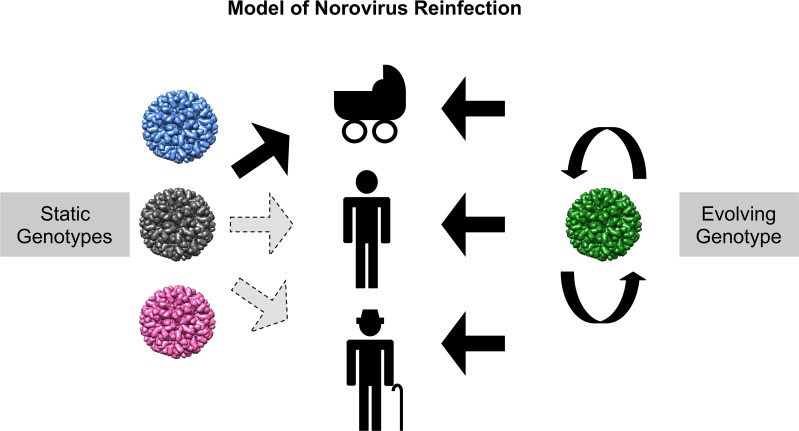
Noroviruses are major pathogens associated with acute gastroenteritis worldwide. Their RNA genomes are diverse, with two major genogroups (GI and GII) comprised of at least 28 genotypes associated with human disease. To elucidate mechanisms underlying norovirus diversity and evolution, we used a large-scale genomics approach to analyze human norovirus sequences. Comparison of over 2000 nearly full-length ORF2 sequences representing most of the known GI and GII genotypes infecting humans showed a limited number (≤5) of distinct intra-genotypic variants within each genotype, with the exception of GII.4. The non-GII.4 genotypes were comprised of one or more intra-genotypic variants, with each variant containing strains that differed by only a few residues over several decades (remaining "static") and that have co-circulated with no clear epidemiologic pattern. In contrast, the GII.4 genotype presented the largest number of variants (>10) that have evolved over time with a clear pattern of periodic variant replacement. To expand our understanding of these two patterns of diversification ("static" versus "evolving"), we analyzed using NGS the nearly full-length norovirus genome in healthy individuals infected with GII.4, GII.6 or GII.17 viruses in different outbreak settings. The GII.4 viruses accumulated mutations rapidly within and between hosts, while the GII.6 and GII.17 viruses remained relatively stable, consistent with their diversification patterns. Further analysis of genetic relationships and natural history patterns identified groupings of certain genotypes into larger related clusters designated here as "immunotypes". We propose that "immunotypes" and their evolutionary patterns influence the prevalence of a particular norovirus genotype in the human population.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Population Genomics of GII.4 Noroviruses Reveal Complex Diversification and New Antigenic Sites Involved in the Emergence of Pandemic Strains.mBio. 2019 Sep 24;10(5):e02202-19. doi: 10.1128/mBio.02202-19. mBio. 2019. PMID: 31551337 Free PMC article.
-
Norovirus genotype diversity associated with gastroenteritis outbreaks in Victoria in 2013.Commun Dis Intell Q Rep. 2015 Mar 31;39(1):E34-41. Commun Dis Intell Q Rep. 2015. PMID: 26063096
-
[Surveillance of norovirus-associated diarrhea in children in Shanghai, 2009-2011].Zhonghua Er Ke Za Zhi. 2014 May;52(5):339-44. Zhonghua Er Ke Za Zhi. 2014. PMID: 24969930 Chinese.
-
Emergence of norovirus strains: A tale of two genes.Virus Evol. 2019 Nov 25;5(2):vez048. doi: 10.1093/ve/vez048. eCollection 2019 Jul. Virus Evol. 2019. PMID: 32161666 Free PMC article. Review.
-
A narrative review of norovirus epidemiology, biology, and challenges to vaccine development.NPJ Vaccines. 2024 May 29;9(1):94. doi: 10.1038/s41541-024-00884-2. NPJ Vaccines. 2024. PMID: 38811605 Free PMC article. Review.
Cited by
-
Molecular Evolution of Human Norovirus GII.2 Clusters.Front Microbiol. 2021 Mar 22;12:655567. doi: 10.3389/fmicb.2021.655567. eCollection 2021. Front Microbiol. 2021. PMID: 33828543 Free PMC article.
-
Novel opportunities for NGS-based one health surveillance of foodborne viruses.One Health Outlook. 2020 Jun 22;2:14. doi: 10.1186/s42522-020-00015-6. eCollection 2020. One Health Outlook. 2020. PMID: 33829135 Free PMC article. Review.
-
Antigenic cartography reveals complexities of genetic determinants that lead to antigenic differences among pandemic GII.4 noroviruses.Proc Natl Acad Sci U S A. 2021 Mar 16;118(11):e2015874118. doi: 10.1073/pnas.2015874118. Proc Natl Acad Sci U S A. 2021. PMID: 33836574 Free PMC article.
-
Identification of a First Human Norovirus CD8+ T Cell Epitope Restricted to HLA-A*0201 Allele.Front Immunol. 2018 Nov 27;9:2782. doi: 10.3389/fimmu.2018.02782. eCollection 2018. Front Immunol. 2018. PMID: 30542352 Free PMC article.
-
Epidemiological, Molecular, and Clinical Features of Norovirus Infections among Pediatric Patients in Qatar.Viruses. 2019 Apr 29;11(5):400. doi: 10.3390/v11050400. Viruses. 2019. PMID: 31035642 Free PMC article.
References
-
- Worobey M, Holmes EC. Evolutionary aspects of recombination in RNA viruses. J Gen Virol. 1999;80 (Pt 10):2535–43. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
