

Telomeres in cancer: tumour suppression and genome instability
- PMID: 28096526
- PMCID: PMC5589191
- DOI: 10.1038/nrm.2016.171
Telomeres in cancer: tumour suppression and genome instability
Erratum in
-
Author Correction: Telomeres in cancer: tumour suppression and genome instability.Nat Rev Mol Cell Biol. 2019 Apr;20(4):259. doi: 10.1038/s41580-019-0113-7. Nat Rev Mol Cell Biol. 2019. PMID: 30816301
Abstract
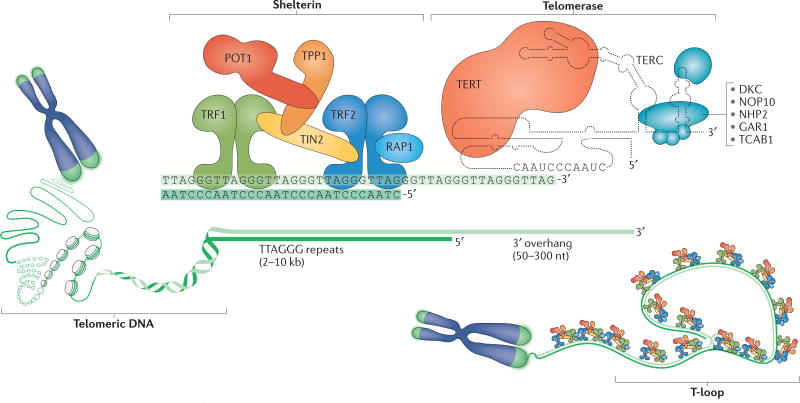
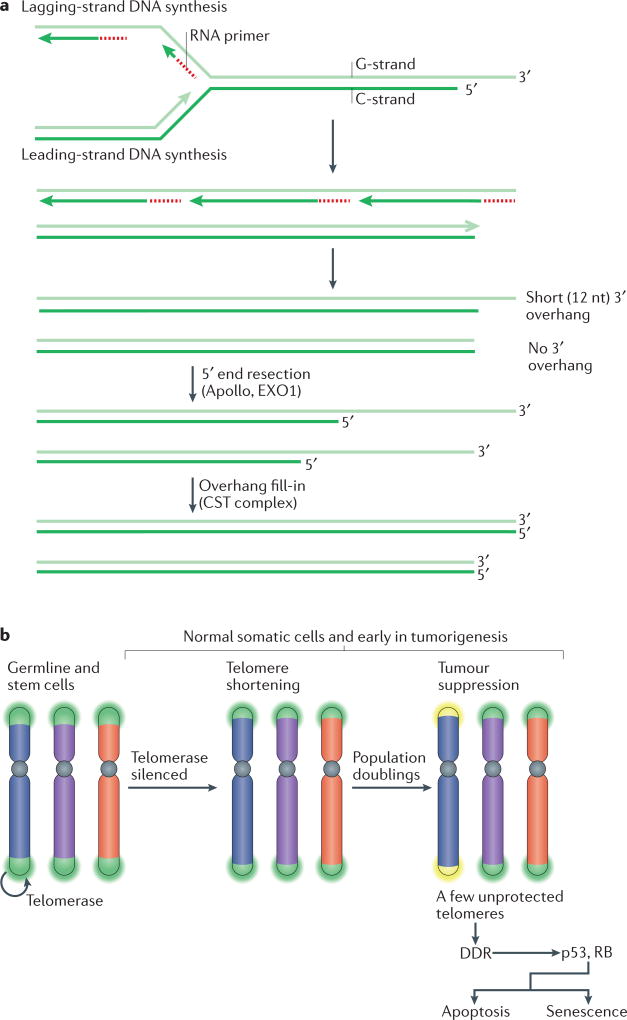
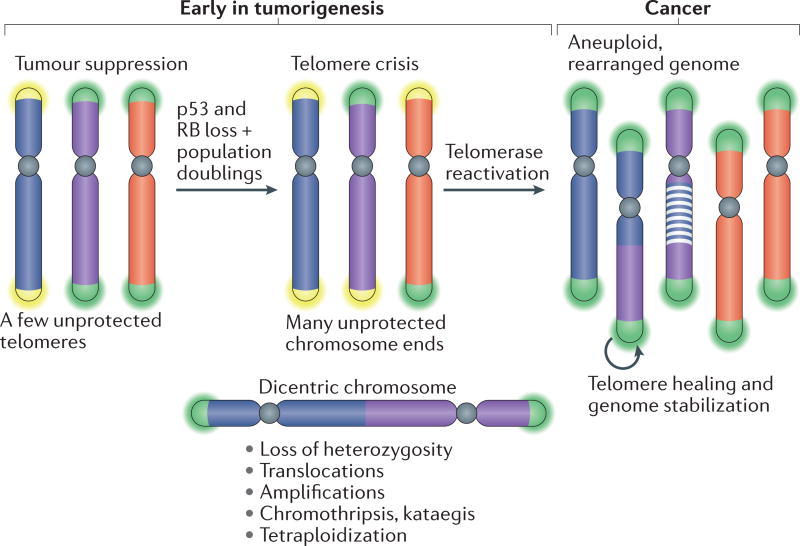
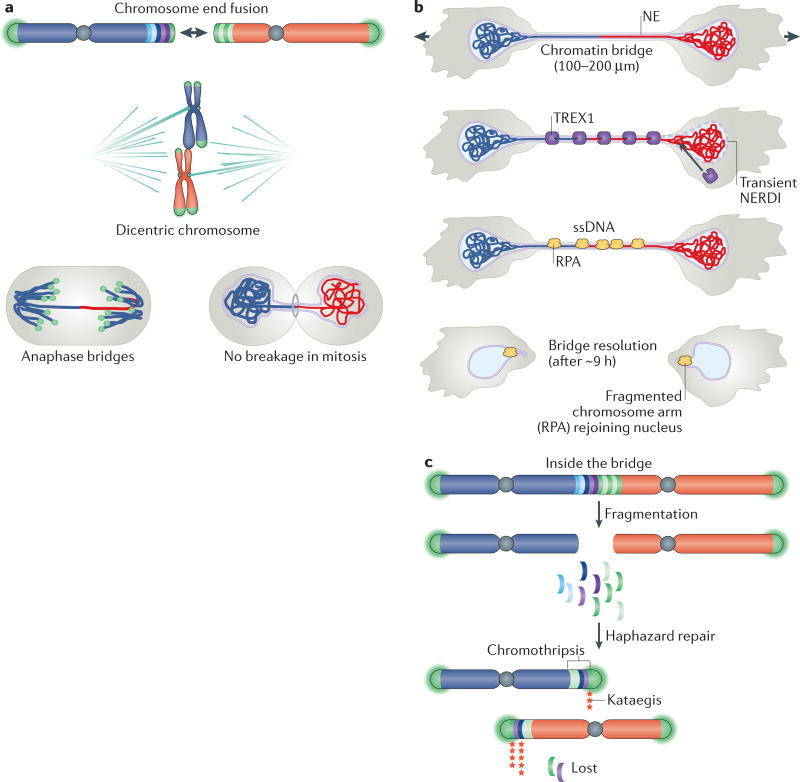
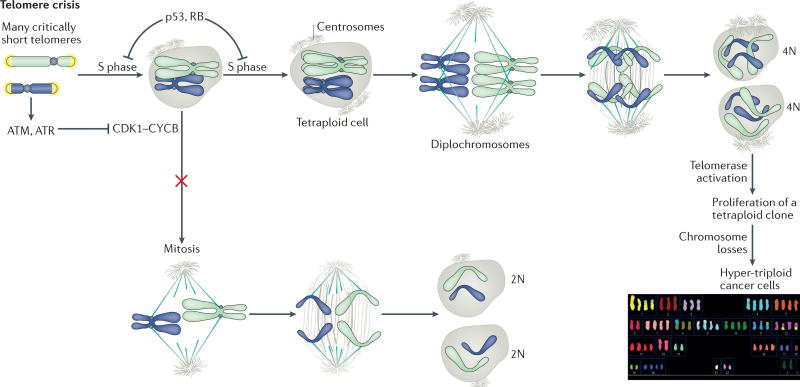
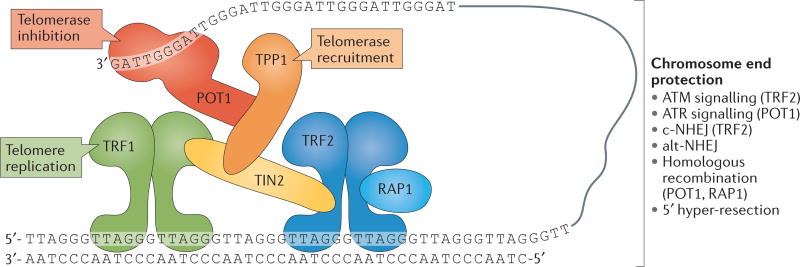
The shortening of human telomeres has two opposing effects during cancer development. On the one hand, telomere shortening can exert a tumour-suppressive effect through the proliferation arrest induced by activating the kinases ATM and ATR at unprotected chromosome ends. On the other hand, loss of telomere protection can lead to telomere crisis, which is a state of extensive genome instability that can promote cancer progression. Recent data, reviewed here, provide new evidence for the telomere tumour suppressor pathway and has revealed that telomere crisis can induce numerous cancer-relevant changes, including chromothripsis, kataegis and tetraploidization.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Telomere Length Dynamics and the Evolution of Cancer Genome Architecture.Int J Mol Sci. 2018 Feb 6;19(2):482. doi: 10.3390/ijms19020482. Int J Mol Sci. 2018. PMID: 29415479 Free PMC article. Review.
-
Telomere instability and cancer.Biochimie. 2008 Jan;90(1):73-82. doi: 10.1016/j.biochi.2007.07.009. Epub 2007 Jul 24. Biochimie. 2008. PMID: 17728038 Review.
-
Biology of telomeres: importance in etiology of esophageal cancer and as therapeutic target.Transl Res. 2013 Dec;162(6):364-70. doi: 10.1016/j.trsl.2013.09.003. Epub 2013 Oct 1. Transl Res. 2013. PMID: 24090770 Free PMC article. Review.
-
APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis.Nat Genet. 2020 Sep;52(9):884-890. doi: 10.1038/s41588-020-0667-5. Epub 2020 Jul 27. Nat Genet. 2020. PMID: 32719516 Free PMC article.
-
Telomeres: Implications for Cancer Development.Int J Mol Sci. 2018 Jan 19;19(1):294. doi: 10.3390/ijms19010294. Int J Mol Sci. 2018. PMID: 29351238 Free PMC article. Review.
Cited by
-
Association of Relative Leucocyte Telomere Length and Gene Single Nucleotide Polymorphisms (TERT, TRF1, TNKS2) in Laryngeal Squamous Cell Carcinoma.Cancer Genomics Proteomics. 2020 Jul-Aug;17(4):431-439. doi: 10.21873/cgp.20202. Cancer Genomics Proteomics. 2020. PMID: 32576588 Free PMC article.
-
Boveri and beyond: Chromothripsis and genomic instability from mitotic errors.Mol Cell. 2024 Jan 4;84(1):55-69. doi: 10.1016/j.molcel.2023.11.002. Epub 2023 Nov 28. Mol Cell. 2024. PMID: 38029753 Free PMC article. Review.
-
Mechanisms of telomere maintenance and associated therapeutic vulnerabilities in malignant gliomas.Neuro Oncol. 2024 Jun 3;26(6):1012-1024. doi: 10.1093/neuonc/noae016. Neuro Oncol. 2024. PMID: 38285162 Free PMC article. Review.
-
TERT Expression in Wilms Tumor Is Regulated by Promoter Mutation or Hypermethylation, WT1, and N-MYC.Cancers (Basel). 2022 Mar 25;14(7):1655. doi: 10.3390/cancers14071655. Cancers (Basel). 2022. PMID: 35406427 Free PMC article.
-
NCBP2 and TFRC are novel prognostic biomarkers in oral squamous cell carcinoma.Cancer Gene Ther. 2023 May;30(5):752-765. doi: 10.1038/s41417-022-00578-8. Epub 2023 Jan 12. Cancer Gene Ther. 2023. PMID: 36635327 Free PMC article.
References
-
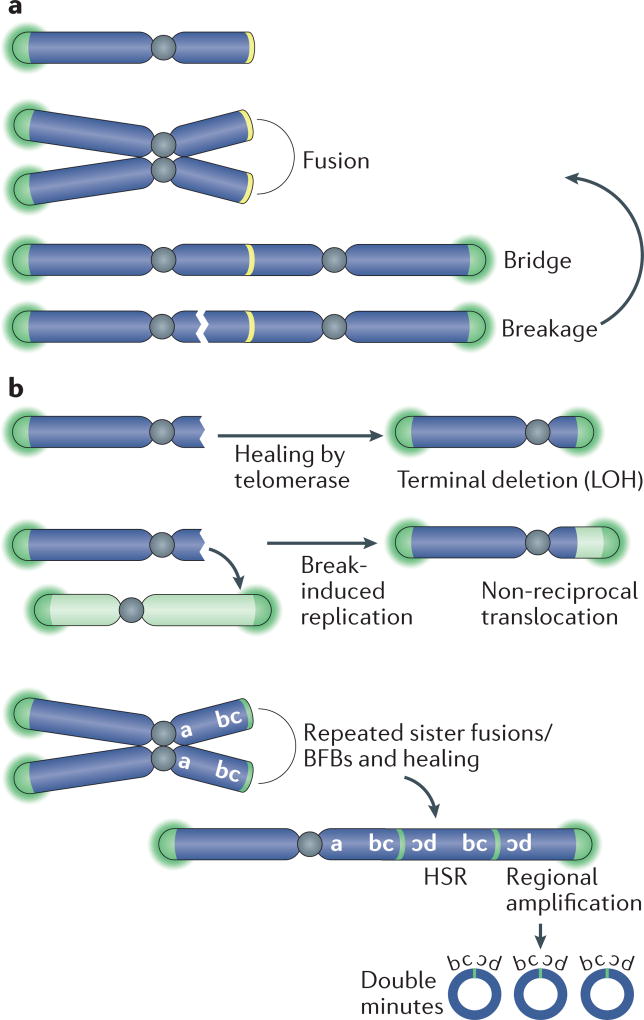
- Artandi SE, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. Demonstrates that telomere attrition in p53-mutant mice promotes epithelial cancers through the formation of chromosome rearrangements. - PubMed
-
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–1654. Shows that dicentric chromosomes formed during telomere crisis persist through mitosis, are fragmented by TREX1 in G1 phase and give rise to chromothripsis and kataegis. - PMC - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
