

Long Noncoding RNA Malat1 Regulates Cerebrovascular Pathologies in Ischemic Stroke
- PMID: 28093478
- PMCID: PMC5320610
- DOI: 10.1523/JNEUROSCI.3389-16.2017
Long Noncoding RNA Malat1 Regulates Cerebrovascular Pathologies in Ischemic Stroke
Abstract
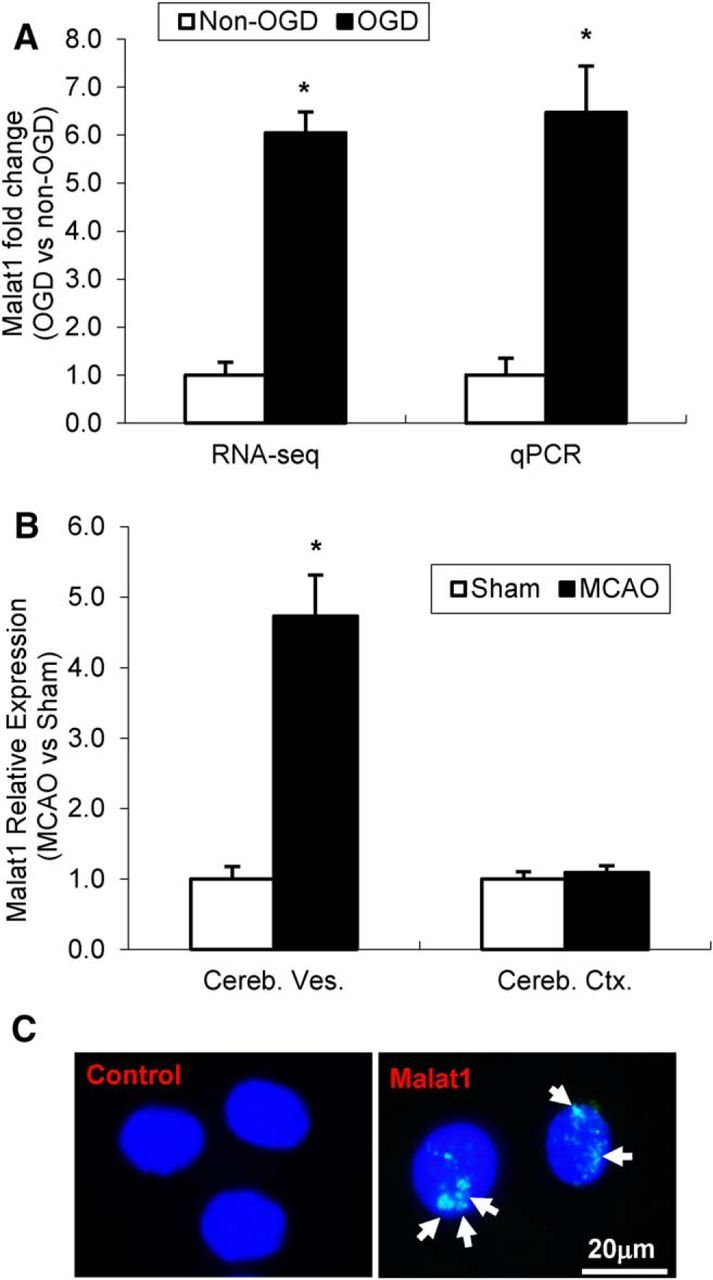
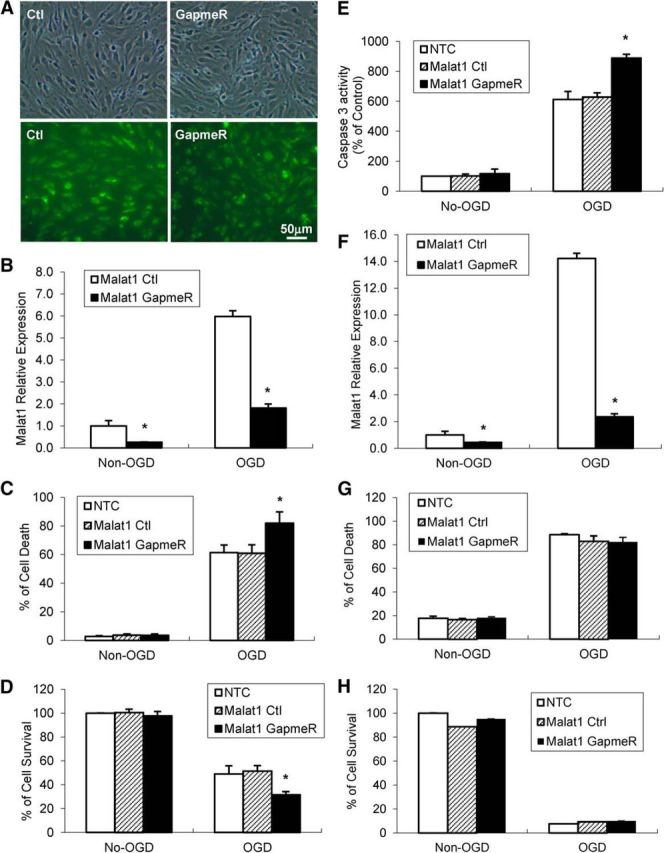
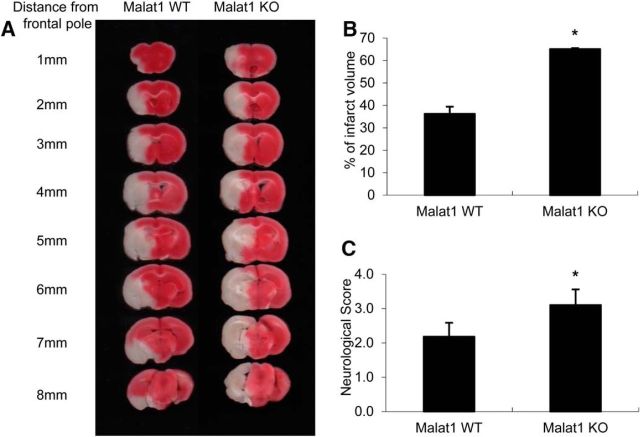
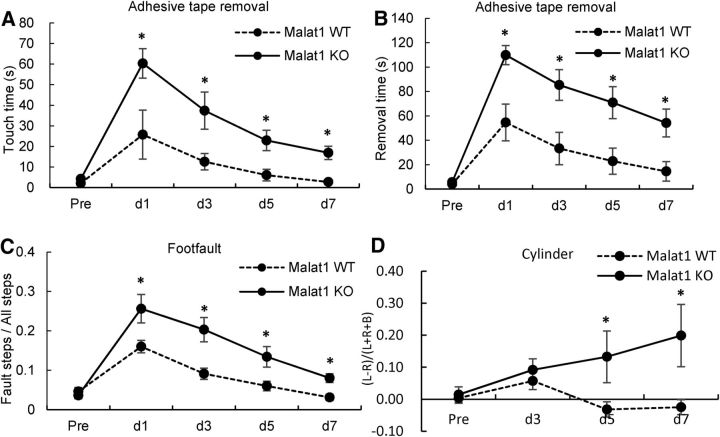
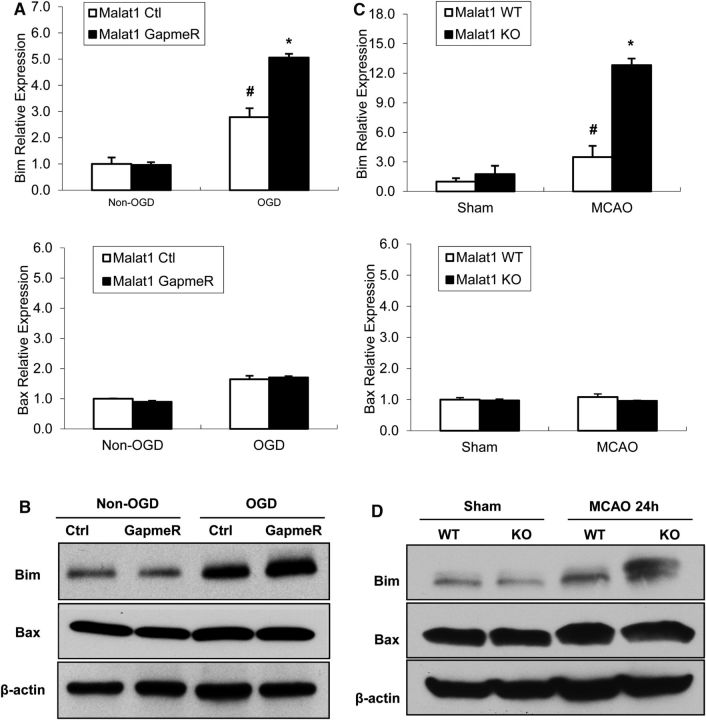
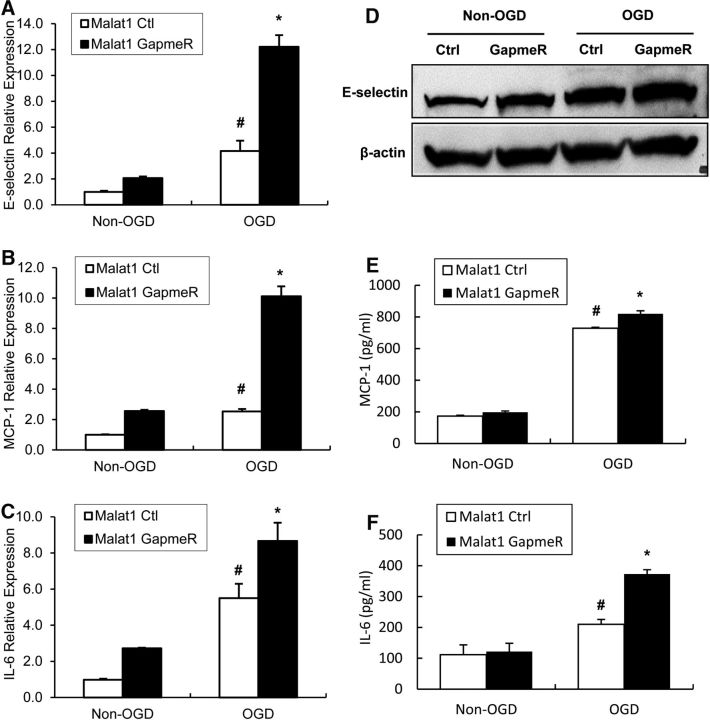
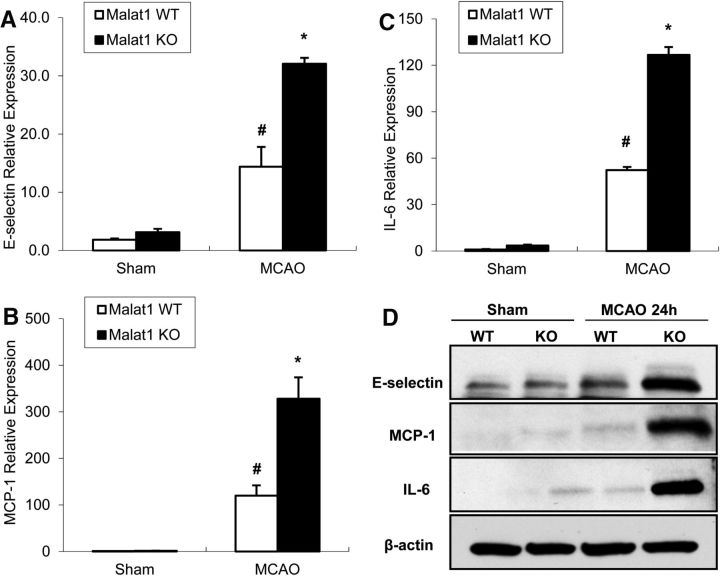
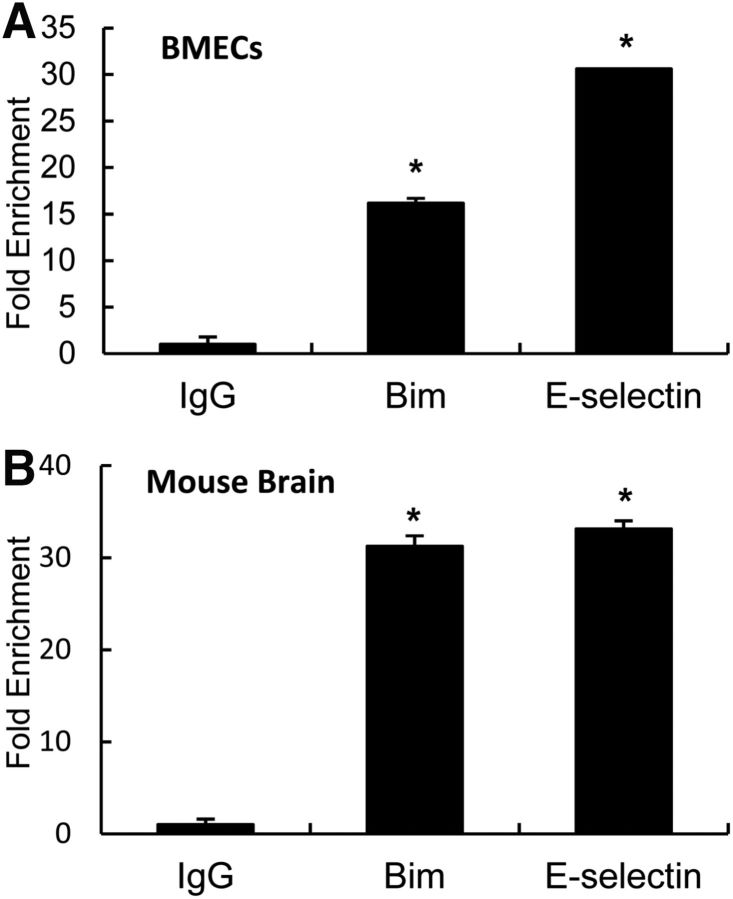
The study was designed to determine the role of long noncoding RNA (lncRNA), metastasis-associated lung adenocarcinoma transcript 1 (Malat1), in ischemic stroke outcome. Primary mouse brain microvascular endothelial cells (BMECs) were cultured and treated with Malat1 GapmeR before 16 h oxygen and glucose depravation (OGD). Cell death was assayed by LDH and MTT methods. Malat1 knock-out and wild-type mice were subjected to 1 h of middle cerebral artery occlusion (MCAO) and 24-72 h of reperfusion. To explore the underlying mechanism, apoptotic and inflammatory factors were measured by qPCR, ELISA, and Western blotting. The physical interaction between Malat1 and apoptotic or inflammatory factors was measured by RNA immunoprecipitation. Increased Malat1 levels were found in cultured mouse BMECs after OGD as well as in isolated cerebral microvessels in mice after MCAO. Silencing of Malat1 by Malat1 GapmeR significantly increased OGD-induced cell death and Caspase 3 activity in BMECs. Silencing of Malat1 also significantly aggravated OGD-induced expression of the proapoptotic factor Bim and proinflammatory cytokines MCP-1, IL-6, and E-selectin. Moreover, Malat1 KO mice presented larger brain infarct size, worsened neurological scores, and reduced sensorimotor functions. Consistent with in vitro findings, significantly increased expression of proapoptotic and proinflammatory factors was also found in the cerebral cortex of Malat1 KO mice after ischemic stroke compared with WT controls. Finally, we demonstrated that Malat1 binds to Bim and E-selectin both in vitro and in vivo Our study suggests that Malat1 plays critical protective roles in ischemic stroke.SIGNIFICANCE STATEMENT Accumulative studies have demonstrated the important regulatory roles of microRNAs in vascular and neural damage after ischemic stroke. However, the functional significance and mechanisms of other classes of noncoding RNAs in cerebrovascular pathophysiology after stroke are less studied. Here we demonstrate a novel role of Malat1, a long noncoding RNA that has been originally identified as a prognostic marker for non-small cell lung cancer, in cerebrovascular pathogenesis of ischemic stroke. Our experiments have provided the first evidence that Malat1 plays anti-apoptotic and anti-inflammatory roles in brain microvasculature to reduce ischemic cerebral vascular and parenchymal damages. Our studies also suggest that lncRNAs can be therapeutically targeted to minimize poststroke brain damage.
Keywords: Malat1; apoptosis; cerebral ischemia; cerebral vascular endothelial cell; inflammation; stroke.
Copyright © 2017 the authors 0270-6474/17/371797-10$15.00/0.
Figures
Similar articles
-
Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia.Exp Neurol. 2016 Mar;277:162-170. doi: 10.1016/j.expneurol.2015.12.014. Epub 2015 Dec 31. Exp Neurol. 2016. PMID: 26746985 Free PMC article.
-
LncRNA MALAT1 up-regulates VEGF-A and ANGPT2 to promote angiogenesis in brain microvascular endothelial cells against oxygen-glucose deprivation via targetting miR-145.Biosci Rep. 2019 Mar 6;39(3):BSR20180226. doi: 10.1042/BSR20180226. Print 2019 Mar 29. Biosci Rep. 2019. Retraction in: Biosci Rep. 2020 Jul 31;40(7):BSR-20180226_RET. doi: 10.1042/BSR-20180226_RET. PMID: 30038058 Free PMC article. Retracted.
-
Down-Regulation of Lncrna MALAT1 Attenuates Neuronal Cell Death Through Suppressing Beclin1-Dependent Autophagy by Regulating Mir-30a in Cerebral Ischemic Stroke.Cell Physiol Biochem. 2017;43(1):182-194. doi: 10.1159/000480337. Epub 2017 Aug 30. Cell Physiol Biochem. 2017. PMID: 28854438
-
A systematic review of the research progress of non-coding RNA in neuroinflammation and immune regulation in cerebral infarction/ischemia-reperfusion injury.Front Immunol. 2022 Oct 7;13:930171. doi: 10.3389/fimmu.2022.930171. eCollection 2022. Front Immunol. 2022. PMID: 36275741 Free PMC article. Review.
-
The long noncoding RNA Malat1: Its physiological and pathophysiological functions.RNA Biol. 2017 Dec 2;14(12):1705-1714. doi: 10.1080/15476286.2017.1358347. Epub 2017 Oct 6. RNA Biol. 2017. PMID: 28837398 Free PMC article. Review.
Cited by
-
Identifying potential therapeutic targets for ischemic stroke through immune infiltration analysis and construction of a programmed cell death‑related ceRNA network.Exp Ther Med. 2022 Sep 21;24(5):680. doi: 10.3892/etm.2022.11616. eCollection 2022 Nov. Exp Ther Med. 2022. PMID: 36185765 Free PMC article.
-
Identification of brain endothelial cell-specific genes and pathways in ischemic stroke by integrated bioinformatical analysis.Brain Circ. 2023 Nov 30;9(4):228-239. doi: 10.4103/bc.bc_40_23. eCollection 2023 Oct-Dec. Brain Circ. 2023. PMID: 38284111 Free PMC article.
-
Identification of Alternative Splicing and LncRNA Genes in Brain Tissues of Fetal Mice at Different Developmental Stages.Comb Chem High Throughput Screen. 2023;26(1):58-82. doi: 10.2174/1386207325666220408091206. Comb Chem High Throughput Screen. 2023. PMID: 35400338
-
LncRNA MALAT1 Promoted Neuronal Necroptosis in Cerebral Ischemia-reperfusion Mice by Stabilizing HSP90.Neurochem Res. 2023 Nov;48(11):3457-3471. doi: 10.1007/s11064-023-03991-z. Epub 2023 Jul 20. Neurochem Res. 2023. PMID: 37470906
-
Targeting of IL-6-Relevant Long Noncoding RNA Profiles in Inflammatory and Tumorous Disease.Inflammation. 2019 Aug;42(4):1139-1146. doi: 10.1007/s10753-019-00995-2. Inflammation. 2019. PMID: 30825076 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous