

The Amyloid Phenomenon and Its Links with Human Disease
- PMID: 28062560
- PMCID: PMC5453392
- DOI: 10.1101/cshperspect.a023648
The Amyloid Phenomenon and Its Links with Human Disease
Abstract
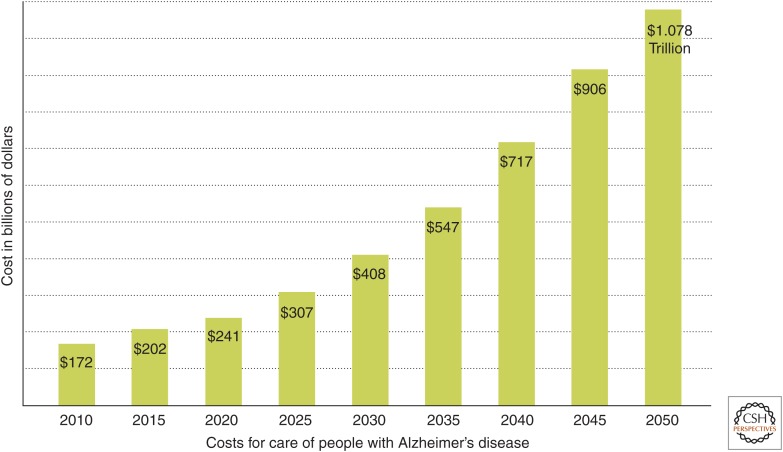
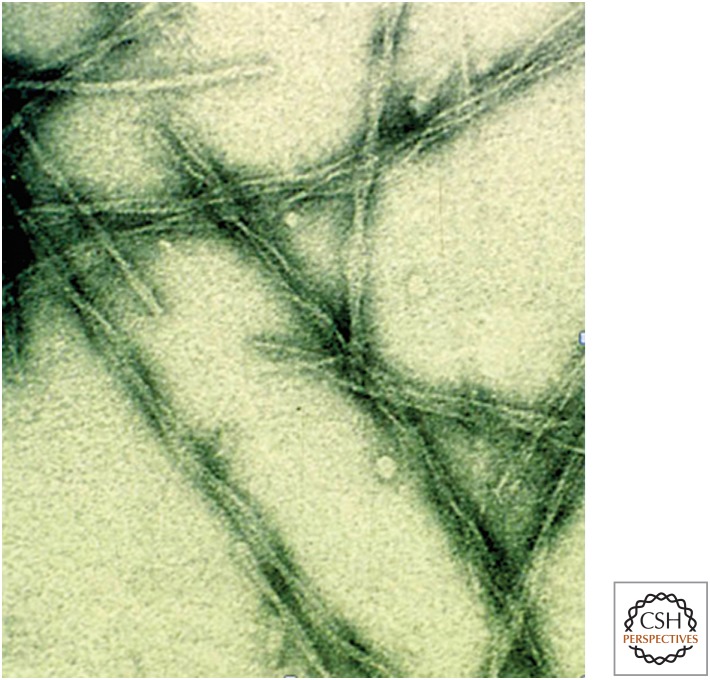
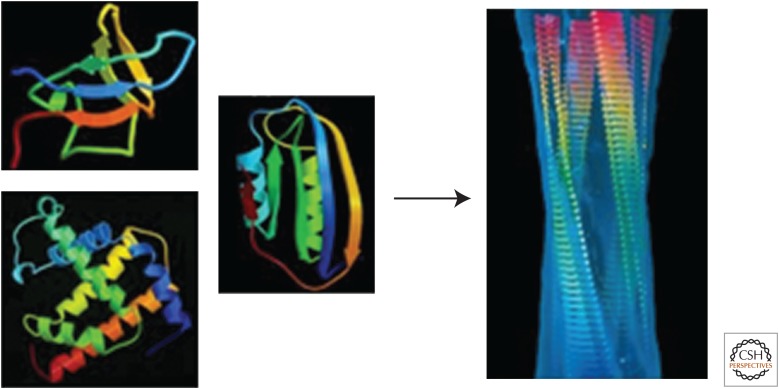
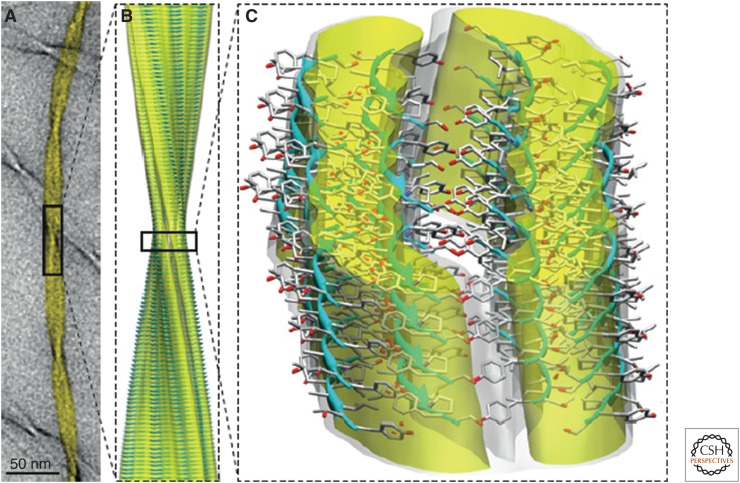
The ability of normally soluble proteins to convert into amyloid fibrils is now recognized to be a generic phenomenon. The overall cross-β architecture of the core elements of such structures is closely similar for different amino acid sequences, as this architecture is dominated by interactions associated with the common polypeptide main chain. In contrast, the multiplicity of complex and intricate structures of the functional states of proteins is dictated by specific interactions involving the variable side chains, the sequence of which is unique to a given protein. Nevertheless, the side chains dictate important aspects of the amyloid structure, including the regions of the sequence that form the core elements of the fibrils and the kinetics and mechanism of the conversion process. The formation of the amyloid state of proteins is of particular importance in the context of a range of medical disorders that include Alzheimer's and Parkinson's diseases and type 2 diabetes. These disorders are becoming increasingly common in the modern world, primarily as a consequence of increasing life spans and changing lifestyles, and now affect some 500 million people worldwide. This review describes recent progress in our understanding of the molecular origins of these conditions and discusses emerging ideas for new and rational therapeutic strategies by which to combat their onset and progression.
Copyright © 2017 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures

Similar articles
-
Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade.Annu Rev Biochem. 2017 Jun 20;86:27-68. doi: 10.1146/annurev-biochem-061516-045115. Epub 2017 May 12. Annu Rev Biochem. 2017. PMID: 28498720 Review.
-
Protein denaturation and aggregation: Cellular responses to denatured and aggregated proteins.Ann N Y Acad Sci. 2005 Dec;1066:181-221. doi: 10.1196/annals.1363.030. Ann N Y Acad Sci. 2005. PMID: 16533927 Review.
-
Fundamentals of cross-seeding of amyloid proteins: an introduction.J Mater Chem B. 2019 Dec 14;7(46):7267-7282. doi: 10.1039/c9tb01871a. Epub 2019 Oct 24. J Mater Chem B. 2019. PMID: 31647489 Review.
-
Theoretical approaches to protein aggregation.Protein Pept Lett. 2006;13(3):287-93. doi: 10.2174/092986606775338407. Protein Pept Lett. 2006. PMID: 16515457 Review.
-
Structure and intermolecular dynamics of aggregates populated during amyloid fibril formation studied by hydrogen/deuterium exchange.Acc Chem Res. 2010 Aug 17;43(8):1072-9. doi: 10.1021/ar9002784. Acc Chem Res. 2010. PMID: 20557067
Cited by
-
Parkinson's Disease: A Comprehensive Analysis of Fungi and Bacteria in Brain Tissue.Int J Biol Sci. 2020 Feb 10;16(7):1135-1152. doi: 10.7150/ijbs.42257. eCollection 2020. Int J Biol Sci. 2020. PMID: 32174790 Free PMC article.
-
Myocilin misfolding and glaucoma: A 20-year update.Prog Retin Eye Res. 2023 Jul;95:101188. doi: 10.1016/j.preteyeres.2023.101188. Epub 2023 May 20. Prog Retin Eye Res. 2023. PMID: 37217093 Free PMC article. Review.
-
Structural Arrangement within a Peptide Fibril Derived from the Glaucoma-Associated Myocilin Olfactomedin Domain.J Phys Chem B. 2021 Mar 25;125(11):2886-2897. doi: 10.1021/acs.jpcb.0c11460. Epub 2021 Mar 8. J Phys Chem B. 2021. PMID: 33683890 Free PMC article.
-
Super-Resolution Infrared Imaging of Polymorphic Amyloid Aggregates Directly in Neurons.Adv Sci (Weinh). 2020 Feb 7;7(6):1903004. doi: 10.1002/advs.201903004. eCollection 2020 Mar. Adv Sci (Weinh). 2020. PMID: 32195099 Free PMC article.
-
A coarse-grained molecular dynamics investigation on spontaneous binding of Aβ1-40 fibrils with cholesterol-mixed DPPC bilayers.Comput Struct Biotechnol J. 2023 Apr 15;21:2688-2695. doi: 10.1016/j.csbj.2023.04.013. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 37143763 Free PMC article.
References
-
- Alzheimer’s Association. 2015. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement 11: 332–384. - PubMed
-
- Apetri MM, Maiti NC, Zagorski MG, Carey PR, Anderson VE. 2006. Secondary structure of α-synuclein oligomers: Characterization by Raman and atomic force microscopy. J Mol Biol 355: 63–71. - PubMed
-
- Arioso P, Vendruscolo M, Dobson CM, Knowles TP. 2014. Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol Sci 35: 127–135. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical