

Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy
- PMID: 28007147
- PMCID: PMC5843375
- DOI: 10.1016/j.jacc.2016.08.079
Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy
Abstract
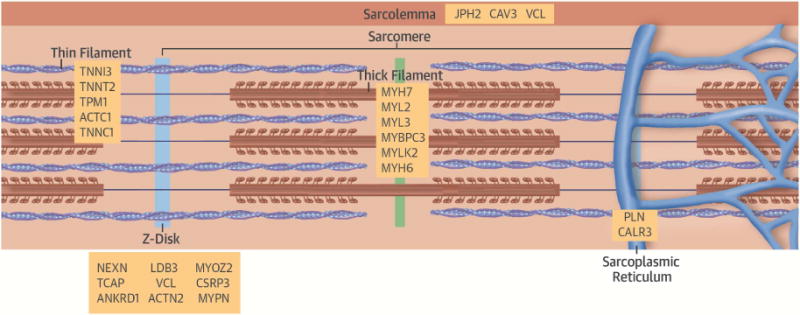
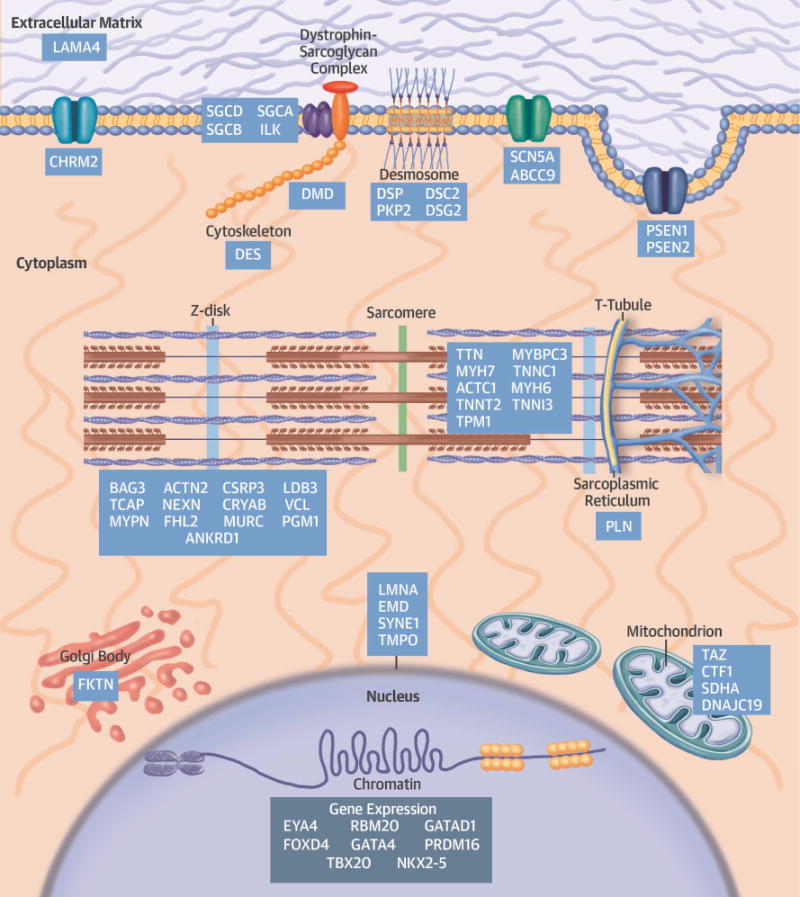
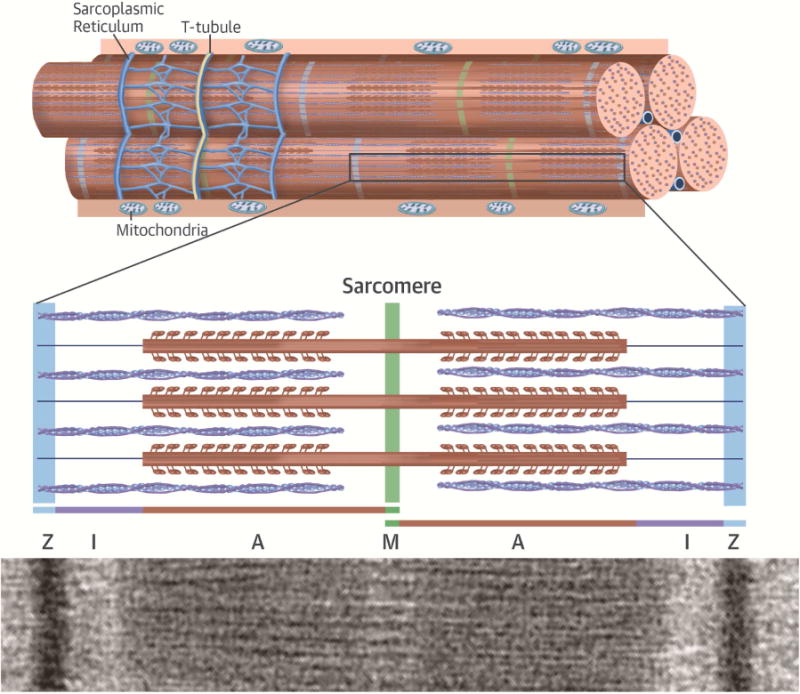
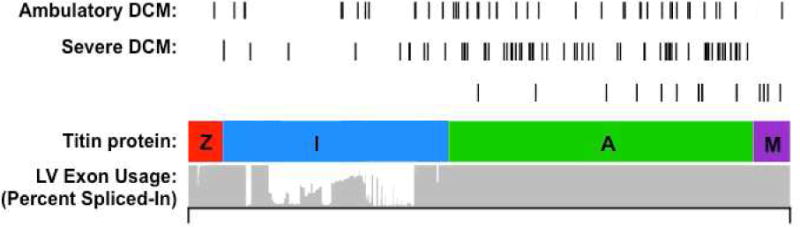
Over the last quarter-century, there has been tremendous progress in genetics research that has defined molecular causes for cardiomyopathies. More than a thousand mutations have been identified in many genes with varying ontologies, therein indicating the diverse molecules and pathways that cause hypertrophic, dilated, restrictive, and arrhythmogenic cardiomyopathies. Translation of this research to the clinic via genetic testing can precisely group affected patients according to molecular etiology, and identify individuals without evidence of disease who are at high risk for developing cardiomyopathy. These advances provide insights into the earliest manifestations of cardiomyopathy and help to define the molecular pathophysiological basis for cardiac remodeling. Although these efforts remain incomplete, new genomic technologies and analytic strategies provide unparalleled opportunities to fully explore the genetic architecture of cardiomyopathies. Such data hold the promise that mutation-specific pathophysiology will uncover novel therapeutic targets, and herald the beginning of precision therapy for cardiomyopathy patients.
Keywords: dilated cardiomyopathy; genetic testing; genetics; hypertrophic cardiomyopathy; molecular etiology; restrictive cardiomyopathy.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Evolving molecular diagnostics for familial cardiomyopathies: at the heart of it all.Expert Rev Mol Diagn. 2010 Apr;10(3):329-51. doi: 10.1586/erm.10.13. Expert Rev Mol Diagn. 2010. PMID: 20370590 Free PMC article. Review.
-
Cardiac Filaminopathies: Illuminating the Divergent Role of Filamin C Mutations in Human Cardiomyopathy.J Clin Med. 2021 Feb 4;10(4):577. doi: 10.3390/jcm10040577. J Clin Med. 2021. PMID: 33557094 Free PMC article. Review.
-
Genetic advances in sarcomeric cardiomyopathies: state of the art.Cardiovasc Res. 2015 Apr 1;105(4):397-408. doi: 10.1093/cvr/cvv025. Epub 2015 Jan 29. Cardiovasc Res. 2015. PMID: 25634555 Free PMC article. Review.
-
Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype.J Am Coll Cardiol. 2007 Mar 27;49(12):1251-64. doi: 10.1016/j.jacc.2006.10.073. Epub 2007 Mar 9. J Am Coll Cardiol. 2007. PMID: 17394955 Review.
-
Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era.J Mol Diagn. 2013 Mar;15(2):158-70. doi: 10.1016/j.jmoldx.2012.09.002. Epub 2012 Dec 27. J Mol Diagn. 2013. PMID: 23274168 Review.
Cited by
-
Sarcomere-based genetic enhancement of systolic cardiac function in a murine model of dilated cardiomyopathy.Int J Cardiol. 2018 Dec 15;273:168-176. doi: 10.1016/j.ijcard.2018.09.073. Epub 2018 Sep 21. Int J Cardiol. 2018. PMID: 30279005 Free PMC article.
-
Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy.Pflugers Arch. 2019 May;471(5):781-793. doi: 10.1007/s00424-018-2226-9. Epub 2018 Nov 20. Pflugers Arch. 2019. PMID: 30456444 Free PMC article. Review.
-
Mitochondrial SLC25 Carriers: Novel Targets for Cancer Therapy.Molecules. 2020 May 22;25(10):2417. doi: 10.3390/molecules25102417. Molecules. 2020. PMID: 32455902 Free PMC article. Review.
-
Genotype-Phenotype Insights of Inherited Cardiomyopathies-A Review.Medicina (Kaunas). 2024 Mar 27;60(4):543. doi: 10.3390/medicina60040543. Medicina (Kaunas). 2024. PMID: 38674189 Free PMC article. Review.
-
Targeted exome analysis of Russian patients with hypertrophic cardiomyopathy.Mol Genet Genomic Med. 2021 Nov;9(11):e1808. doi: 10.1002/mgg3.1808. Epub 2021 Oct 1. Mol Genet Genomic Med. 2021. PMID: 34598319 Free PMC article.
References
-
- Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation. 2006;113:1807–16. - PubMed
-
- Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–6. - PubMed
-
- Jarcho JA, McKenna W, Pare JA, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–8. - PubMed
-
- Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants : a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
