

Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS)
- PMID: 28003278
- PMCID: PMC5538408
- DOI: 10.1101/cshperspect.a024117
Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS)
Abstract
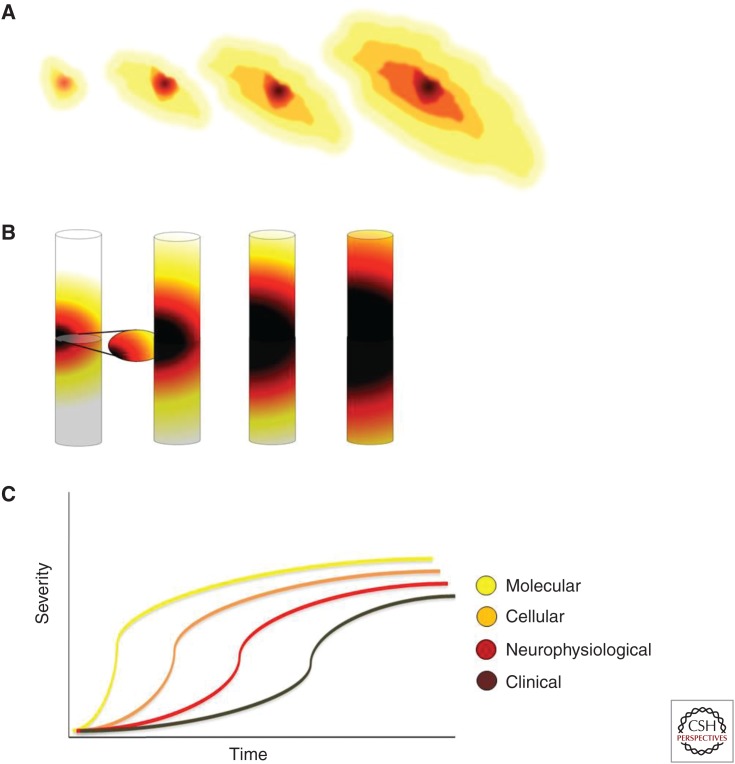
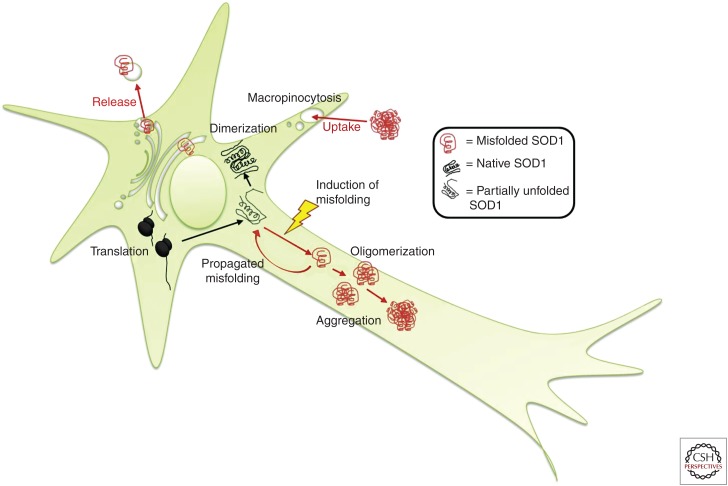
Amyotrophic lateral sclerosis (ALS) is primarily characterized by progressive loss of motor neurons, although there is marked phenotypic heterogeneity between cases. Typical, or "classical," ALS is associated with simultaneous upper motor neuron (UMN) and lower motor neuron (LMN) involvement at disease onset, whereas atypical forms, such as primary lateral sclerosis and progressive muscular atrophy, have early and predominant involvement in the UMN and LMN, respectively. The varying phenotypes can be so distinctive that they would seem to have differing biology. Because the same phenotypes can have multiple causes, including different gene mutations, there may be multiple molecular mechanisms causing ALS, implying that the disease is a syndrome. Conversely, multiple phenotypes can be caused by a single gene mutation; thus, a single molecular mechanism could be compatible with clinical heterogeneity. The pathogenic mechanism(s) in ALS remain unknown, but active propagation of the pathology neuroanatomically is likely a primary component.
Copyright © 2017 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures

Similar articles
-
Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis.Amyotroph Lateral Scler Frontotemporal Degener. 2013 May;14 Suppl 1(0 1):5-18. doi: 10.3109/21678421.2013.778548. Amyotroph Lateral Scler Frontotemporal Degener. 2013. PMID: 23678876 Free PMC article.
-
Regional spreading pattern is associated with clinical phenotype in amyotrophic lateral sclerosis.Brain. 2023 Oct 3;146(10):4105-4116. doi: 10.1093/brain/awad129. Brain. 2023. PMID: 37075222 Free PMC article.
-
Uncovering amyotrophic lateral sclerosis phenotypes: clinical features and long-term follow-up of upper motor neuron-dominant ALS.Amyotroph Lateral Scler. 2011 Jul;12(4):278-82. doi: 10.3109/17482968.2011.580849. Amyotroph Lateral Scler. 2011. PMID: 21702734
-
Emerging understanding of the genotype-phenotype relationship in amyotrophic lateral sclerosis.Handb Clin Neurol. 2018;148:603-623. doi: 10.1016/B978-0-444-64076-5.00039-9. Handb Clin Neurol. 2018. PMID: 29478603 Review.
-
Amyotrophic lateral sclerosis; clinical features, differential diagnosis and pathology.Int Rev Neurobiol. 2024;176:1-47. doi: 10.1016/bs.irn.2024.04.011. Epub 2024 May 22. Int Rev Neurobiol. 2024. PMID: 38802173 Review.
Cited by
-
Free energy calculations of ALS-causing SOD1 mutants reveal common perturbations to stability and dynamics along the maturation pathway.Protein Sci. 2021 Sep;30(9):1804-1817. doi: 10.1002/pro.4132. Epub 2021 Jun 22. Protein Sci. 2021. PMID: 34076319 Free PMC article.
-
Unlocking the therapeutic potential of P2X7 receptor: a comprehensive review of its role in neurodegenerative disorders.Front Pharmacol. 2024 Jul 30;15:1450704. doi: 10.3389/fphar.2024.1450704. eCollection 2024. Front Pharmacol. 2024. PMID: 39139642 Free PMC article. Review.
-
The double-edged functions of necroptosis.Cell Death Dis. 2023 Feb 27;14(2):163. doi: 10.1038/s41419-023-05691-6. Cell Death Dis. 2023. PMID: 36849530 Free PMC article. Review.
-
P2X7 receptor activation mediates superoxide dismutase 1 (SOD1) release from murine NSC-34 motor neurons.Purinergic Signal. 2022 Dec;18(4):451-467. doi: 10.1007/s11302-022-09863-5. Epub 2022 Apr 28. Purinergic Signal. 2022. PMID: 35478453 Free PMC article.
-
Metabolic Abnormalities, Dietary Risk Factors and Nutritional Management in Amyotrophic Lateral Sclerosis.Nutrients. 2021 Jun 30;13(7):2273. doi: 10.3390/nu13072273. Nutrients. 2021. PMID: 34209133 Free PMC article. Review.
References
-
- Abe K, Fujimura H, Toyooka K, Sakoda S, Yorifuji S, Yanagihara T. 1997. Cognitive function in amyotrophic lateral sclerosis. J Neurol Sci 148: 95–100. - PubMed
-
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. 2011. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 122: 691–702. - PubMed
-
- ALS Mutation Database. 2007. The University of Tokyo; (reseq.biosciencedbc.jp/resequence/SearchDisease.do?targetId=1).
-
- Andres PL, Thibodeau LM, Finison LJ, Munsat TL. 1987. Quantitative assessment of neuromuscular deficit in ALS. Neurol Clin 5: 125–141. - PubMed
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602–611. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous