

DNA methylation in the gene body influences MeCP2-mediated gene repression
- PMID: 27965390
- PMCID: PMC5206576
- DOI: 10.1073/pnas.1618737114
DNA methylation in the gene body influences MeCP2-mediated gene repression
Abstract
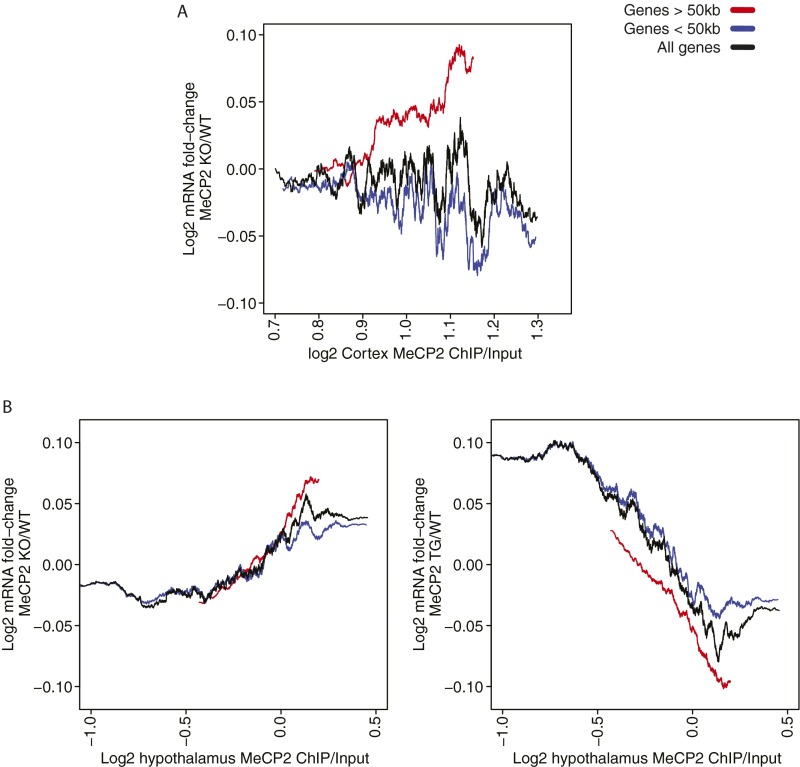
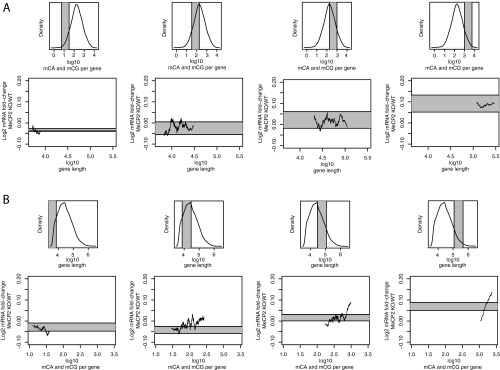
Rett syndrome is a severe neurodevelopmental disorder caused by mutations in the methyl-CpG binding protein gene (MECP2). MeCP2 is a methyl-cytosine binding protein that is proposed to function as a transcriptional repressor. However, multiple gene expression studies comparing wild-type and MeCP2-deficient neurons have failed to identify gene expression changes consistent with loss of a classical transcriptional repressor. Recent work suggests that one function of MeCP2 in neurons is to temper the expression of the longest genes in the genome by binding to methylated CA dinucleotides (mCA) within transcribed regions of these genes. Here we explore the mechanism of mCA and MeCP2 in fine tuning the expression of long genes. We find that mCA is not only highly enriched within the body of genes normally repressed by MeCP2, but also enriched within extended megabase-scale regions surrounding MeCP2-repressed genes. Whereas enrichment of mCA exists in a broad region around these genes, mCA together with mCG within gene bodies appears to be the primary driver of gene repression by MeCP2. Disruption of methylation at CA sites within the brain results in depletion of MeCP2 across genes that normally contain a high density of gene-body mCA. We further find that the degree of gene repression by MeCP2 is proportional to the total number of methylated cytosine MeCP2 binding sites across the body of a gene. These findings suggest a model in which MeCP2 tunes gene expression in neurons by binding within the transcribed regions of genes to impede the elongation of RNA polymerase.
Keywords: DNA methylation; MeCP2; Rett syndrome; transcription.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





Similar articles
-
Disruption of DNA-methylation-dependent long gene repression in Rett syndrome.Nature. 2015 Jun 4;522(7554):89-93. doi: 10.1038/nature14319. Epub 2015 Mar 11. Nature. 2015. PMID: 25762136 Free PMC article.
-
MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes.Mol Cell. 2020 Jan 16;77(2):294-309.e9. doi: 10.1016/j.molcel.2019.10.032. Epub 2019 Nov 26. Mol Cell. 2020. PMID: 31784358 Free PMC article.
-
MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome.Proc Natl Acad Sci U S A. 2015 Apr 28;112(17):5509-14. doi: 10.1073/pnas.1505909112. Epub 2015 Apr 13. Proc Natl Acad Sci U S A. 2015. PMID: 25870282 Free PMC article.
-
DNA methylation and Rett syndrome.Hum Mol Genet. 2003 Oct 15;12 Spec No 2:R221-7. doi: 10.1093/hmg/ddg286. Epub 2003 Aug 19. Hum Mol Genet. 2003. PMID: 12928486 Review.
-
Regulation mechanism and research progress of MeCP2 in Rett syndrome.Yi Chuan. 2014 Jul;36(7):625-30. doi: 10.3724/SP.J.1005.2014.0625. Yi Chuan. 2014. PMID: 25076025 Review.
Cited by
-
Decoding the Epigenetic Landscape: Insights into 5mC and 5hmC Patterns in Mouse Cortical Cell Types.bioRxiv [Preprint]. 2024 Jul 10:2024.07.06.602342. doi: 10.1101/2024.07.06.602342. bioRxiv. 2024. PMID: 39026756 Free PMC article. Preprint.
-
Toxicity of overexpressed MeCP2 is independent of HDAC3 activity.Genes Dev. 2018 Dec 1;32(23-24):1514-1524. doi: 10.1101/gad.320325.118. Epub 2018 Nov 21. Genes Dev. 2018. PMID: 30463906 Free PMC article.
-
Hypermethylation of PDX1, EN2, and MSX1 predicts the prognosis of colorectal cancer.Exp Mol Med. 2022 Feb;54(2):156-168. doi: 10.1038/s12276-022-00731-1. Epub 2022 Feb 15. Exp Mol Med. 2022. PMID: 35169223 Free PMC article.
-
MeCP2: A Critical Regulator of Chromatin in Neurodevelopment and Adult Brain Function.Int J Mol Sci. 2019 Sep 16;20(18):4577. doi: 10.3390/ijms20184577. Int J Mol Sci. 2019. PMID: 31527487 Free PMC article. Review.
-
Rett syndrome: insights into genetic, molecular and circuit mechanisms.Nat Rev Neurosci. 2018 Jun;19(6):368-382. doi: 10.1038/s41583-018-0006-3. Nat Rev Neurosci. 2018. PMID: 29740174 Free PMC article. Review.
References
-
- Chahrour M, Zoghbi HY. The story of Rett syndrome: From clinic to neurobiology. Neuron. 2007;56(3):422–437. - PubMed
-
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27(3):327–331. - PubMed
-
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27(3):322–326. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
